reSee.it - Tweets Saved By @KevinMcCairnPhD

@KevinMcCairnPhD - Kevin W. McCairn PhD
https://bmj.com/content/344/bmj.e2398/rapid-responses https://gab.com/Flavinkins/posts/109663743902085653 You can only have GoF work generate a viable prediction or a vaccine if the next emergence is of the exact same genetic makeup as your GOF strain. The extreme diversity of viruses mean that you have (total number of viruses in the world (billions)*probability that a GOF study result being used maliciously (>1/200 minimum)) times higher likelihood that GOF research cause a pandemic in stead of preventing it. https://archive.ph/BToZR https://archive.md/YYIXp


@KevinMcCairnPhD - Kevin W. McCairn PhD
There were never a single viable vaccine or therapeutic ever created from GOF research.
@KevinMcCairnPhD - Kevin W. McCairn PhD
There have never been a single productive prevention move (none claimed by their reports have been implemented), not a single working therapeutic or vaccine (no currently used antivirals or vaccine came out of EHA funded research), made by the EHA. The EHA did GOF after GOF and grew virus after virus. No currently fielded vaccine, therapeutic or preventative originated from GOF research.
@KevinMcCairnPhD - Kevin W. McCairn PhD
All currently fielded vaccines were developed using circulating strains in the targeted species (e.g. humans). GOFROC or virus hunting are wholly unnecessary for any vaccines or therapeutics. And unfortunately, the first use of the prefusion-stabilized Spike was to target MERS-CoV and WIV1 wasn’t the drive of the patent, nor did the patent required any GOF research. Also, the vaccines are now proven to be hazardous, driving unending waves of reinfections.



@KevinMcCairnPhD - Kevin W. McCairn PhD
One of the reasons why GOF virology need closed ranks is because they have to hide the fact that their actions are not only catastrophically dangerous, they are also not productive at all in any positive ways. https://archive.ph/BToZR https://archive.md/YYIXp https://gab.com/Flavinkins/posts/108828145093274513
@KevinMcCairnPhD - Kevin W. McCairn PhD
There have never been a single productive prevention move (none claimed by their reports have been implemented), not a single working therapeutic or vaccine (no currently used antivirals or vaccine came out of EHA funded research), made by the EHA. The EHA did GOF after GOF and grew virus after virus.



@KevinMcCairnPhD - Kevin W. McCairn PhD
No currently fielded vaccine, therapeutic or preventative originated from GOF research.
@KevinMcCairnPhD - Kevin W. McCairn PhD
Their “preventive measures”? Never implemented. Their “vaccine and therapeutic”? None even reached the point of receiving a name. Their Bioweapons operations under the name of “understanding XXX virus spillover potential”? Too many that couldn’t even be counted with hands and feet.
@KevinMcCairnPhD - Kevin W. McCairn PhD
And they have never ever prevented any pandemics, produced any viable vaccines or therapeutics, or produced any constructive results in the reduction of any of the subjects they claimed to tracking.
@KevinMcCairnPhD - Kevin W. McCairn PhD
ePPP virologists and virus hunters are arsonists that pretends to be firefighters, and they have only ever seeked to weaponize the pathogens they “tracks” with the pretext of dual-use “research” without ever coming up with any actual clinically executed effort in their alleged goal of “prevention and treatment” for any of the pathogens.
@KevinMcCairnPhD - Kevin W. McCairn PhD

@KevinMcCairnPhD - Kevin W. McCairn PhD
All they get are epidemics and pandemics. No vaccine, therapeutic, or preventions that have reached production have ever yielded from any of these risky research. https://gab.com/Flavinkins/posts/109523507275843148
@KevinMcCairnPhD - Kevin W. McCairn PhD
https://en.wikipedia.org/wiki/2007_United_Kingdom_foot-and-mouth_outbreak Fact: Pirbright was not shut down after FMDV leak infecting 4 farms nearby. They repaired their drain pipes and continued operation, not even interfering academic publication patterns. Ironically, cow farms were shut down and beef trade was closed during the outbreak. This resulted in an epidemic lasting 5 months in cows that lead to at least two major cullings and severe disruption to the livestock trade from the U.K. The reaction look like what they claim a zoonosis would look like, not what they declare what the WIV would do when a shut-down would certainly directly admit guilt and spell doom to both the institute and its operators. https://www.theaustralian.com.au/science/beijing-lab-mishap-infected-scientist-with-covid19/news-story/9b0cb0ed84df21d25da11b698be3611a Fact 2: there is no shut-down reported at all in the IVDC either after the 2004 leak of SARS or the 2020 leak of Covid. Not even a burp of interruption. Fact 3: the WIV went on hiatus to the bat CoV isolation tests over 2020-2023. During the sverdlovsk anthrax leak, they blamed the animal farms and markets nearby and did not officially shut down the facility. The construction of another anthrax facility nearby was considered potential indication of a shut-down, which is on par with the WIV hiatus. After a timescale similar to the WIV hiatus, the new facility was opened for inspection which no anthrax was found, meaning that they fixed sverdlovsk and went on, just like the WIV (chen WEI……). In fact, there have not been a single record of an lab leak or LAI in a research facility that resulted in the (especially permanent, as what they claimed would happen) shut down of the facility (despite hundreds of known incidents in record), even with significant epidemic resulted. (Ebola21, FMDV07, H1N177, Anthrax82 which no official shutdown was known).

@KevinMcCairnPhD - Kevin W. McCairn PhD
Offshoring of risky virological lab work to countries with lower or zero biosafety requirements is an old strategy. Lassa/Ebola lab, Kenema Government Hospital, Sierra Leone. https://reuters.com/article/us-bioterror-africa-idUKTRE71D49820110214/ https://who.int/news-room/feature-stories/detail/the-last-ebola-survivor-of-his-team- https://ncbi.nlm.nih.gov/pmc/articles/PMC5050470/pdf/jiw239.pdf http://vhfc.org Ebola at BSL-2. https://ncbi.nlm.nih.gov/pmc/articles/PMC5050470/ https://ncbi.nlm.nih.gov/pmc/articles/PMC5050470/bin/supp_214_suppl-3_S110__index.html Supplementary Figure 1A. 2014-2021 Ebola. It is nearly universal for outbreaks to be traced back far before their official site of discovery.




@danwalker9999 - Daniel A. Walker 🇨🇦🇺🇦🇬🇱🌻😷💉🚴🏻
@emilyakopp Offshoring of risky virological lab work to countries with lower or zero biosafety requirements is an old strategy. Lassa/Ebola lab, Kenema Government Hospital, Sierra Leone. https://www.reuters.com/article/us-bioterror-africa-idUKTRE71D49820110214/ https://www.who.int/news-room/feature-stories/detail/the-last-ebola-survivor-of-his-team- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5050470/pdf/jiw239.pdf http://vhfc.org



@KevinMcCairnPhD - Kevin W. McCairn PhD
Metabiota and USAID ePPP “research” have led to also the RVFV outbreak in the nile river.

@KevinMcCairnPhD - Kevin W. McCairn PhD
Notice how not only were measures that were taken after known lab leaks being identical to that is claimed “for zoonosis”, cover-up efforts especially in authoritarian regimes Also involves the distortion and adulteration of the officially provided “datasets” to push “zoonosis” when a lab or a bioweapons facility leaks.
@KevinMcCairnPhD - Kevin W. McCairn PhD
Which is also why they preliminarily banned criticizing of the official narrative regarding the market or mentioning about the Wuhan lab before the outbreak was or can be announced.


@KevinMcCairnPhD - Kevin W. McCairn PhD
https://arxiv.org/abs/2109.09112 No. GOF don’t make us safer. Nipah is a regionally self limiting disease. Any GOF over it are intended to change this property. Any culturing outside their endemic regions lead to unnatural epidemics which will never happen if it was not conducted. https://gab.com/Flavinkins/posts/109523507275843148
@KevinMcCairnPhD - Kevin W. McCairn PhD
The cover-up of the soviet union was a single letter away from that of communist china.
@KevinMcCairnPhD - Kevin W. McCairn PhD
With all of the information that can come out of China being under control of the communist party, it is expected that manipulation of the scientific process and research using intentionally adulterated and fabricated “data” and “releases” will be performed by the communists—to push the origin away from their labs based on western behavior—which invoked essentially the identical playbook as the Soviet Union in the 1979 sverdlovsk anthrax leak, over comprable timeframes.

@KevinMcCairnPhD - Kevin W. McCairn PhD

@KevinMcCairnPhD - Kevin W. McCairn PhD
@DrLiMengYAN1 @LawrenceSellin Authoritarian regimes uses lies regularly, and “science” was among the fields which they lie with.
@FilippaLentzos - Dr Filippa Lentzos
Matt Meselson’s wonderful closing words: “We should teach our students that once you’re a scientist you have a public responsibility, not just to work in your laboratory, but if you see some good you can do outside of your lab, think about whether you can do it, maybe you should”
@kakape - Kai Kupferschmidt
In April 1979 dozens of people died in the deadliest anthrax outbreak on record. What happened in the city of Sverdlovsk? A new @pandemiapodcast episode on anthrax, bioweapons and the search for the truth is out. Full episode (in 🇩🇪) here: https://viertausendhertz.de/pan49/ Thread to come:
@KevinMcCairnPhD - Kevin W. McCairn PhD
@DrLiMengYAN1 @LawrenceSellin @DrJBhattacharya @NIHDirector_Jay
@KevinMcCairnPhD - Kevin W. McCairn PhD
@DrLiMengYAN1 @LawrenceSellin @DrJBhattacharya @NIHDirector_Jay GOFROC is the equivalent of underaged children inserting 🧨 into piles of 💩 to see the explosion—it provided zero benefits to society except for the pleasure of its perpetrators.

@KevinMcCairnPhD - Kevin W. McCairn PhD
In fact, ePPP GOFROC frequently damage vaccine research by producing strains that lacked immune evasion—which in turn lead to vaccine candidates (when they claim to make one) which work only on the particular ePPP GOFROC product and no other strains (and only briefly). They fail in the real world unless said product was released.
@KevinMcCairnPhD - Kevin W. McCairn PhD
Of course, biological attack agents and GOFROC strains are frequently outsourced in their development to evade scrutiny and often to push some of the risks away to other locations—usually under a “dual use” umbrella, that always make no progress in the civilian field nor result in any civilian use in reality.
@KevinMcCairnPhD - Kevin W. McCairn PhD
In many cases, ePPP GOFROC don’t bother to propoose any civilian uses at all. No solutions given, only dangerous strains that satisfies the perverse desire of the “researchers” with a potential for weaponization for mass destruction—zero benefit, only harm.
@zerohedge - zerohedge
South Korea Lab Makes Bird Flu 100% Lethal In Mammals: 'Virology Journal' https://www.zerohedge.com/political/south-korea-lab-makes-bird-flu-100-lethal-mammals-virology-journal

@KevinMcCairnPhD - Kevin W. McCairn PhD
https://x.com/nestcommander/status/1781826378683556254?s=46&t=wRQSWp_1VffWmS2vKQwhSA… When they begun an official censorship campaign on all and anything on the “Wuhan P4 lab”, @AP you know that they are acting out a pre-scripted cover-up effort, and was not “genuingly clueless”. Especially when the expected precaution in response to an ongoing outbreak, such as washing hands, was also intentionally avoided. https://x.com/daoyu15/status/1718782597114016231?s=46&t=wRQSWp_1VffWmS2vKQwhSA… The entirety of the “they covered up the market” theory can also be interpreted as that they don’t want to let the NEGATIVE animal samples from being known. They need to shroud it in mystery, or the https://x.com/daoyu15/status/daoyu15/status/1694163822473629792?s=46&t=wRQSWp_1VffWmS2vKQwhSA… absence of any secondary spillovers in any other markets in China https://x.com/nestcommander/status/1779485262005023009?s=46&t=wRQSWp_1VffWmS2vKQwhSA…, when SARS1 have already did 7 times over the same month and a half of no actions https://x.com/daoyu15/status/daoyu15/status/1687891376665681920, would be confirmed in the total absence of any positive animal swabs at the market. https://x.com/daoyu15/status/daoyu15/status/1754661054733242856 China, as in the national level, performed sampling of the entirety of the wildlife supply chain toward the market and have officially insisted that “it came from illegal wildlife sold at the Huanan market” up to May 2020. They even tried to blame pangolins, among the others. All the sampling results are negative, and which are in fact leaked even during this period of “you can blame only the animals”. https://x.com/daoyu15/status/daoyu15/status/1668828125617352704?s=46&t=wRQSWp_1VffWmS2vKQwhSA… And once again, a lower down that was tasked to eliminate potential negative evidence by the higher up, but only told to eliminate “potential evidence” in general, can easily come up with conspiracy theories about “they are tasking to eliminate positive evidence”; https://x.com/nestcommander/status/1775081708007878978?s=46&t=wRQSWp_1VffWmS2vKQwhSA… When in fact, The fear that a thorough investigation would turn up confirming all animals being negative and that their labs would become blamed immediately, being the the real cause, is evident in both the absence of spillovers anywhere alongside or in any other destinations of the wildlife supply chain, and the leaked preliminary sampling efforts among these supply chains that returned no positivity at all, https://x.com/daoyu15/status/1740641874032185732?s=46&t=wRQSWp_1VffWmS2vKQwhSA… Despite at the time when “wild animals illegally traded at the Huanan market” and then “wild animals” was the only permitted origin theory on all official outlets in China (up to May 2020).
@KevinMcCairnPhD - Kevin W. McCairn PhD
When they begun an official censorship campaign on all and anything on the “Wuhan P4 lab”, you know that they are acting out a pre-scripted cover-up effort, and was not “genuingly clueless”. Especially when the expected precaution in response to an ongoing outbreak, such as washing hands, was also intentionally avoided.


@KevinMcCairnPhD - Kevin W. McCairn PhD
And of course, they also attempted to blame it on a smuggled animal, or frozen food, as the negativity of all wildlife sampling (which is already known in Jan-Feb 2020 with the data leaks archive.md/iw1Pz archive.md/4rVph on failing to get positive results even in the upstream supply farms of the market) in China is confirmed by total absence of secondary outbreaks or secondary spillovers in China when raccoon dogs have already been made livestock and wildlife trade being still alive and well online. Their evident tampering of the early cases databases And their blatant and statistically unsound lies over the serology of all the cases before the market outbreak (which none are linked to any wildlife markets at all) Confirmed that their agenda is to push the outbreak onto the market nomatter what source they have to blame on, even if they have to disclose the negativity of tests as leaked before, they need to find an explanation as “frozen food or smuggled animal”.
@KevinMcCairnPhD - Kevin W. McCairn PhD
Before they begun enforcing their claim of “100/174 centered around the market” and starting to tamper with data to make the claim, https://ghrp.biomedcentral.com/articles/10.1186/s41256-021-00200-8 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7149375/ 135/92 and 115/82 cases already got into in early peer-reviewed papers that went missing in the WHO report. Past media reports archive.md/Ea0Kw archive.md/1x658 also contradict WHO in key early cases’ residences, including the earliest case they admit in the WHO report. archive.md/5sdkR archive.md/1pcCU archive.md/N0hib archive.md/VXtu9 archive.is/Kyr1z https://archive.org/details/mace-e-pai-covid-19-analysis-redacted/page/8/mode/1up And you know that they hate this information when it was censored. The MACE-EPAI document here is not searchable on google. Up to one third of all cases were either removed completely or moved toward the market in the “dataset”. archive.md/zUD1F archive.md/Pc6gp https://archive.is/p3K3Z Including the very first case they ever admitted officially. And outright removed 4 times more cases than official. Unlinked cases supposedly secondary to linked cases should cluster around them, not the market itself. archive.md/GvRcD archive.md/ZgVzp Wuhan authorities after that archive.md/OIGPz 2014 incident now targeted only the Huanan market when looking for EID outbreaks—and nowhere else. archive.md/1x658 They tampered with the early cases data archive.md/Ea0Kw To make it look like it “started at the market” when in reality the first case they ever admitted lived right next to the WIV BSL-4. archive.md/5sdkR severe discrepancy happening December 2019 and January 2020 indicate tampering with case counts. archive.md/1pcCU This is indicative of catastrophic ascertainment bias was going on. None of China’s “early cases” dataset is credible. https://archive.md/ET1GA https://archive.md/Ea0Kw https://archive.md/1x658 The tampering of early case residence data is systematic and extensive. It is the reason why they refused to provide this data in any detail at all. Not only did The first every case they admitted live in Shidong right next to the BSL-4, and were moved toward the market in the WHO report in contradiction to all known media coverage, https://gab.com/Flavinkins/posts/109256201942085712 the entirety of Wuchang district was wiped clean for every single WHO case that have onset before 27/12/2019–with up to 3000 cases moved to the market this way over the entire Wuhan outbreak. https://archive.md/1x658 and for central Wuchang near the labs and the densest inhabited regions inside the district, all cases were moved away in the WHO map. Unfortunately Rasmussen's work on the origins question rests heavily on what David Relman described as "hopelessly impoverished" early case data. https://www.washingtonpost.com/national-security/2023/02/27/little-known-scientific-team-behind-new-assessment-covid-19-origins/ https://www.washingtonpost.com/opinions/2022/11/17/covid-early-cases-wuhan-china-mystery/ https://archive.md/ke1lp https://archive.md/RaYPC David Fisman: I think the most interesting thing this fellow says is that there are clearly tens of thousands of cases...That implies a much earlier introduction than would have occurred with a seafood market outbreak..."






@KevinMcCairnPhD - Kevin W. McCairn PhD
https://archive.md/UIBkB https://archive.md/6LuXg https://web.archive.org/web/20231101133202/https://en.rattibha.com/thread/1718570491534061745 archive.md/ZgVzp https://archive.md/fWTg1 The Huanan market was the only place in Wuhan monitored for EID since 2014. This is to ensure that whenever an outbreak occurs the only place it will be first detected would be at the Huanan market itself, ensuring that all lab escapes can be covered up and scapegoat campaigns can initiate to blame the wildlife trade in stead. The official narrative is always that “it was caused by wild animals sold in the Huanan market” and “It have an origin within illegally traded wild animals” all the way up to May 2020–the pub date for the Pangolin papers is up to April 2020 and that a bounty to “find the animal origins” for SARS-CoV-2 is still active in May 2020. The “most likely origin being wild animals” argument is present in Chinese articles about SARS-CoV-2 all over 2020, where numerous attempts at finding animal hosts were performed in-vitro but always land onto Homo Sapiens and leave their “primary suspects” otherwise being animals not sold at the Huanan market, to much of the dismay of the authors, despite the themes being almost always “there is a broad host range for SARS-CoV-2” to try stretch the search efforts as wide as possible, as long as possible, bidding to keep up with the increasingly longer list of negative farms and species in the national search efforts. Despite their numerous attempts at removing the human correlational edge with SARS-CoV-2, their effort ultimately failed with either the proportionality and thus the unique positive correlation and mutual information between Homo Sapiens and SARS-CoV-2 is preserved, or with all of the mutual information between SARS-CoV-2 and any land-dwelling species at all being destroyed.




@KevinMcCairnPhD - Kevin W. McCairn PhD
https://michaelweissman.substack.com/p/an-inconvenient-probability-v57 The meter-precise market centered KDE of W+P “unlinked cases” required in addition to unreasonable and knowingly false assumptions on the social contact and shared space structures of market cases including visitors especially when they occur outside the market, an total absence of anisotropies and biases within the populational and movement structures around the market, neither of which are proven, and quite the opposite—an extreme level of anisotropy, again biasing toward where the linked cases are found, are observed. Yet somehow the unlinked cases were ~50m farther away in opposite to these bias directions when compared to the market itself, which once again required precise recentering of “data” by manipulative actions. (edited) An Inconvenient Probability v5.7 Bayesian analysis of the probable origins of Covid. Quantifying "friggin' likely"
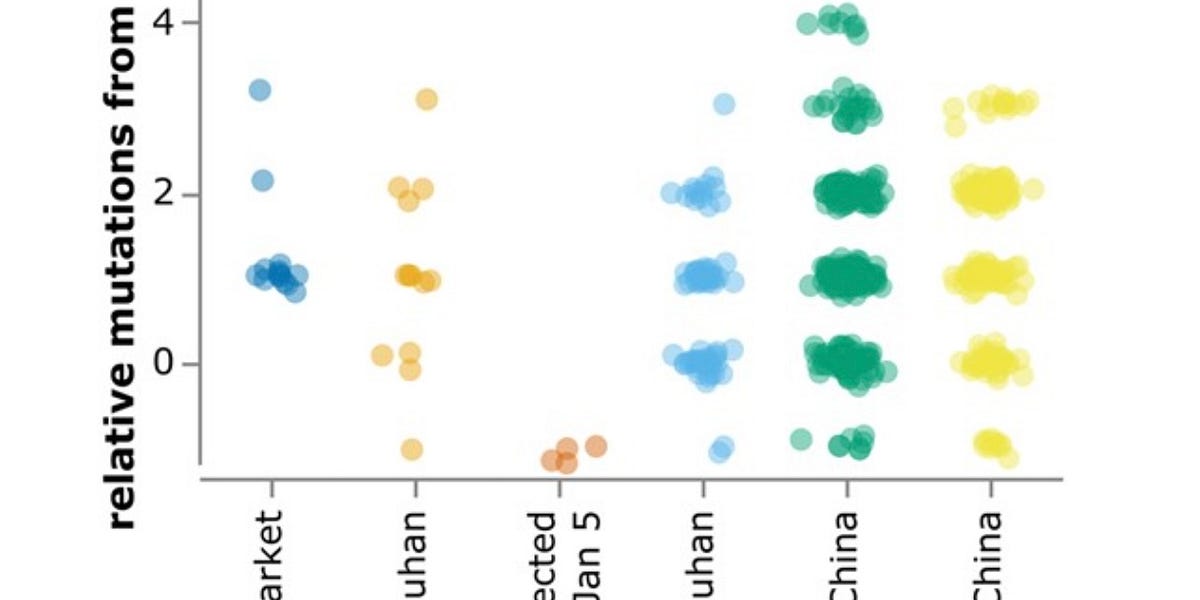
@KevinMcCairnPhD - Kevin W. McCairn PhD
To the earliest Wuhan authorities that prioritize “blame the animals” to prevent scrutiny to the lab (which they also happen to initiate before any public inquiries on this matter could even begin), “no swabs” are better than “negative swabs”. Sadly some of these animal swabs do got taken away at that time just because “gather relevant samples as close as possible and then inspect them” being one of the standard procedures for many of the agencies-of-interest outside Wuhan, and when they are examined, again, per standard procedures, the results got leaked in January 2020, and all of which are negative.
@NestCommander - Kevin W. McCairn PhD
https://x.com/daoyu15/status/1867436175415554170?s=46&t=wRQSWp_1VffWmS2vKQwhSA… Of course. “TBD” for a year and a half despite already had all the experimental material requirements from the sourcing genome to the testing set-ups, and have already completed the majority of the tests a year and half before, is indicative of an inconvenient negative result that needs hiding and guarantee the next publication to be deepfake.
@NestCommander - Kevin W. McCairn PhD
https://www.sciencedirect.com/science/article/pii/S2666389922001039 @COVIDSelect @Rebecca21951651 @ban_epp_gofroc @BiophysicsFL @DrLiMengYAN1 @lude_media @FBI @mattwridley @msabouri @LawrenceSellin @WashburneAlex @quay_dr @BillyBostickson Just remember: since about late August 2024, both China and the west have completed the construction of AI based methodologies related to generative adversarial networks(GANs) to falsify scientific data. (And obviously you can use transcriptome mapping methods to carve animal and viral transcriptomes into fraudulent “samples” before merging them.) We should not trust any of the “data” from conflicted virologists in any way now.


@NestCommander - Kevin W. McCairn PhD
@NestCommander - Kevin W. McCairn PhD
And of course. The full native gene and specific conditions inside an animal is what that matters for infectivity. https://docs.google.com/document/d/1HeZdCnDA4WpoO_kzISjmoFzIAqgFha3fNEPvBpp2z3M/edit https://docs.google.com/document/d/18d_IMZU_DYRX1DlXuSNySC_4DiTcgabjhxoz9K4tWrg/edit That is why in practice the actual observed host range of SARS-CoV-2 is much, much narrower than the claims of “broad host tropism”, and which all old world wildlife species when their native habitats overlap with that of a rhinolophus bat that carries an ACE2-using Sarbecovirus, failed to become infected in nature by any virus that carried the SARS-CoV-2 RBD including SARS-CoV-2 itself.

@NestCommander - Kevin W. McCairn PhD
https://t.co/S2eU226ILx They already had the genome assemblies required to source all the announced “TBD” genes available in 2019-2020 and which the extraction have already been demonstrated from these exact genomes. They already had and have completed the majority of the tests that they wanted to do with the exact formats and protocols of the tests all fully finished in mid 2023. And with a conflict so great that their livelihoods and heads depends on the uncontrolled and continued funding of GOF research, any “results” they would create or release, extending from this exact test that was already a year and a half old, would definitely be faked. The methodologies would include but not be specific to: sample switch-outs, protocol switch-outs, subject switch-outs, AI mediated fabrication of data and images, alignment-based pick and replace for sequencing results.
@NestCommander - Kevin W. McCairn PhD
https://t.co/aJ3P9XphhP (Which they specified in their 2023 report that they sourced their genes for testing via searching through published genome assemblies and sequences using sequence alignment, and which this particular method have been demonstrated to be used for the extraction of one of their TBD sequences by another publication in 2023 from an 2019 assembly.).
@NestCommander - Kevin W. McCairn PhD
These kind of fake science especially the later ones that are based on diffusion models, are impossible to spot. This was from china’s case-specific trained GAN model (e.g. too early in technology that it can be disclosed especially as a proof-of-concept to hint out their western zoonati colluders. Deboosted on Google and can be found only through Bing). The ones above are from the west where the model used was a generalist (e.g. off-the-shelf) diffusion model with the right prompt (context-conditioned infill/outpainting). They can essentially craft whatever species/sample/complex data as they wanted to push for their colluded intended conclusion by using what that are available to them (but failed to provide support to their colluded intended conclusion) as conditioning.


@NestCommander - Kevin W. McCairn PhD
https://sciencedirect.com/science/article/pii/S2666389922001039 @COVIDSelect @Rebecca21951651 @ban_epp_gofroc @BiophysicsFL @DrLiMengYAN1 @lude_media @FBI @mattwridley @msabouri @LawrenceSellin @WashburneAlex @quay_dr @BillyBostickson Just remember: since about late August 2024, both China and the west have completed the construction of AI based methodologies related to generative adversarial networks(GANs) to falsify scientific data. (And obviously you can use transcriptome mapping methods to carve animal and viral transcriptomes into fraudulent “samples” before merging them.) We should not trust any of the “data” from conflicted virologists in any way now.

@NestCommander - Kevin W. McCairn PhD
And of course. The full native gene and specific conditions inside an animal is what that matters for infectivity. https://docs.google.com/document/d/1HeZdCnDA4WpoO_kzISjmoFzIAqgFha3fNEPvBpp2z3M/edit https://docs.google.com/document/d/18d_IMZU_DYRX1DlXuSNySC_4DiTcgabjhxoz9K4tWrg/edit That is why in practice the actual observed host range of SARS-CoV-2 is much, much narrower than the claims of “broad host tropism”, and which all old world wildlife species when their native habitats overlap with that of a rhinolophus bat that carries an ACE2-using Sarbecovirus, failed to become infected in nature by any virus that carried the SARS-CoV-2 RBD including SARS-CoV-2 itself.
@NestCommander - Kevin W. McCairn PhD
https://t.co/3rrwEoR08a “science” is extremely susceptible to the tampering and poisoning of the underlying “data” by authoritarian regimes. Including with inconsistencies and artifacts that forensically invalidates the “datasets”. And of course, methodologies for data fraud especially from the CCP have obviously kept improving over time.
@NestCommander - Kevin W. McCairn PhD
Only the Homo Sapiens would be real in any newer posts from these conflicted “virologists”. https://t.co/TF5dZbUWLE


@NestCommander - Kevin W. McCairn PhD
https://gab.com/Flavinkins/posts/109221415649435463 https://gab.com/Flavinkins/posts/109255356915252021 During the construction of full-length genomes by DEFUSE, the actual methodology is “consensus sequence construction”—e.g. they gather multiple sequences, usually a pool of reads, and then during the construction process, reads (and SNVs) are selected based on whether it can generate the correct site for the cloning process, in addition to the usual consensus generation process, where “unique mutations” from batches of QS are specifically singled out, and, if judged to be driven by a likely sequencing error, removed. The introduction of random designed single nucleotide changes to introduce the necessary site would effectively undo the intended utility of consensus construction, where “sufficiently supported” reads from multiple genomes or multiple reads in SRA (note the “Yunnan bats” libraries were constructed and sequenced in mid 2019) are assembled with the constraint of generating an readily clonable genome, and which one to several alternative test sequences (generate and evaluate until 3-5 viable sequences are obtained each year) are evaluated per pool of genomes and reads utilized. This consensus construction process is constrained by the need to generate a pattern that is easy to clone, and to prevent introducing additional “sequencing errors”, the specific sites are always picked from existing sequences e.g. those that are used in the construction of the consensus. If you just pick a random mutation there is a major risk that (especially in the ORF1ab) the RNA structure is broken and the result is not viable, which is in fact one of the reasons why they want to clone consensus sequences out of sample databases and Short read sequencing data in the full-length QS validation tests, in the first place.




@NestCommander - Kevin W. McCairn PhD
https://x.com/daoyu15/status/daoyu15/status/1731414539324018732 CAS “pathogen host adaptation and immune intervention” is one of such major grants that they continued DEFUSE on. https://x.com/daoyu15/status/daoyu15/status/1747929537848266981 https://x.com/daoyu15/status/1748424486729253321





@NestCommander - Kevin W. McCairn PhD
Politically significant data are state secrets in China. There is no probability that they would remain untampered when released by China. web.archive.org/web/2023110113… archive.md/UODyy archive.md/kJDII archive.md/g2L31 archive.md/Z72Mb Lies and cover-up of China proves intent of https://web.archive.org/web/20231024174245/https://en.rattibha.com/thread/1714851707719668064 creating and releasing SARS-CoV-2. https://web.archive.org/web/20231029094650/https://en.rattibha.com/thread/1717097760942354718 A20, B5, Q61, inconsistency and inconsistency in the China “Huanan market” data and anomalies in data release times indicate significant tampering of these politically significant “data”archive.md/luOy6 archive.md/ryr5p https://resee.it/tweet/1714851707719668064 https://archive.md/2mQwP It is authoritarian takeover. The CCP and the WEF are both responsible for releasing and then covering up the origin for SARS-CoV-2. All of the “Huanan market” data cases or environmental samples have been tampered with. https://archive.md/VYCRK https://web.archive.org/web/20231024174245/https://en.rattibha.com/thread/1714851707719668064 Catastrophic data inconsistency and anomalies in outbreak response by China clearly indicate intent in deliberate creation and intentional release of SARS-CoV-2. https://archive.md/79xvI None of their “data” are trustable in any way. There in fact is an intent to fabricate data. https://archive.md/bUVYp https://archive.md/26Y3G The inconsistencies upon inconsistencies and problems upon problems of Chinese “data”. Tamper with data, and you get caught. And all of the “data” you post loses all credibility. In fact, all pro-zoonosis datasets from China have been tampered with. The key problems here is simply that absence proper versioning or custody of “data” they put up, on GISAID or in the WHO report, archive.md/0aHWr https://archive.md/Myt4u there is no credibility at all in any piece of “data” China made. @DrLiMengYAN1 archive.md/52DyQ archive.md/B0xlW https://web.archive.org/web/20231101133202/https://en.rattibha.com/thread/1718570491534061745 archive.md/kJDII archive.md/UODyy archive.md/g2L31 archive.md/Z72Mb archive.md/AjLkp archive.md/luOy6 archive.md/ryr5p A20, B5, Q61, inconsistency and inconsistency in the China “Huanan market” data and anomalies in data release times indicate significant tampering of these politically significant “data”.




@NestCommander - Kevin W. McCairn PhD
Nearly all of the early bat and pangolin datasets including the critical RaTG13 amplicon datasets have been changed multiple times before or just as they were being published, day after delay as they change them according to requirements as fast as possible. Trust China for ANY origin-relevant “data” now? @washburnealex @humblesci B5, A20, F13, F46, Q61, none of the politically significant “data” of China can be trusted in any way. And the most likely origin of the “positive wildlife stall samples” in the first place, is the WCDC and the AMMS in Wuhan planted them under the command of the bioweapons program—and did a poor job doing so. They even covered up the fact that humans can shed the virus and blocked it from the national IVDC and CCDC until the aforementioned institutions have to visit Wuhan hospitals themselves and find out that human to human transmission can in fact occur. Tamper with data, and you get caught. And all of the “data” you post loses all credibility. All pro-zoonosis “early sample datasets” in China have been tampered with. Bad badgers in Q61, Inverse correlation between human*SARS-CoV-2 and total 300nt+ mammalian contigs in the samples The “market samples” dataset is just as tampered with and with clear artifacts left behind, as all the “animal origin” datasets uploaded by China previously. A20, B5, Q61, every single inconsistency within the “market dataset” directly implicate tampering of and therefore non-validity of the “data”.




@NestCommander - Kevin W. McCairn PhD
Real forensic evidence never get an explanation of official release from China. All China show you as “forensic evidence” have already been tampered with. Not just Ascertainment bias. The “data” have been actively tampered with. Why bleach the toilets before sampling it? Not only there were no gloves at all in the A20 stall W7-15-17, but the sample is both inconsistent in viral reads between 2021/2023 and in host fractions (despite claimed to be multiplex PCR directly from the original sample which should not change host fractions) between the lineage reads-free “metagenomic” and the lineage A “viral amplicons” datasets. Like all 26/03/2023 deposition date “datasets”, they were tampered beyond credibility (3/4 of all samples with an 2021 viral count have been changed, the 1 left was kept without any distinction indicate likely used as standard and debunking the attempted explanation of resequencing (all 4 are stated to be “resequenced via multiplex PCR” together) by contradiction (an unchanged B5 requires an 2021 resequencing date, F13/F54/A20 moved to resemble B5 in ratios in 2023 clearly a move to cover-up https://archive.md/ANS4Q the prior artifacts that became known when they begun monitoring relevant online info in March 2023). Not only Jan 12 samples were affected—for all sampling dates mutual information with all species have been destroyed with the scrambling of the host reads upon the inclusion of the 26/03/2023 upload date datasets. It is quite evident that removing contigs does not eliminate Correlation between humans and the virus because reads are removed proportionally. And all that scrambling just removed mutual information to all species, in all sampling dates. And still can not establish significant mutual information to any “susceptible species”. All zoonosis datasets in China have been tampered with, again artifacts are seen. As found out by NCBI FOIA. https://t.co/WOsGj2kqFZ@washburnealex Both of the PCoV BioProjects with changed DBs change them so that they can remove human DNA from the samples. (One left VERO bits behind, one removed so many sequences that viral concentration in the “metagenomes” were 50x-70x higher than their virus culture.) No explicit Chinese DBs on origin can be trusted in any way. Unfortunately, they failed to consider that there are alternative assemblies and non-standard hypervariable regions in humans though. arxiv.org/abs/2108.08163 arxiv.org/abs/2207.03288 Tamper with data, and you get caught. And all of the “data” you post loses all credibility. x.com/drlimengyan1/s… As for the reason why they need to fabricate the bat and pangolin data as they do with (A20, F13, F46)/B5 and Q61? x.com/drlimengyan1/s… They release a fallback to animal whenever the lab is scrutinized beyond denial. Another issue here for GISAID, is always that archive.md/0aHWr https://archive.md/Myt4u there is no credibility at all due to absence of proper versioning or custody of “data” they put up. archive.md/52DyQ archive.md/B0xlW @DiLiMengYAN1




@NestCommander - Kevin W. McCairn PhD
Chen lived in Shidong. Even by the annexes indicating his history. The only thing they did is that they moved him to Jianghan close to the market on the WHO maps. Also, Chen is not the only person infected in Shidong/Jiangxia and central Wuchang. Most were censored and only one of the two ambulances arriving in 31/12/2019 have been registered as a dot—likely because the origin wasn’t inside the Shidong prefecture/BSL-4 surroundings, and likely only because of being a close contact relative of Chen (contacting an known case). Chen’s accidental inclusion in the WCH’s first report of early cases and its subsequent media coverage mean that China have no choice but to tamper with the official data in an attempt to move him—while the HPHICWM attempt to whistleblow the “cluster 1” cases in 26-27/12/2019 generated from the WCDC’s leak of their culture stock (intended for sample manipulation) was blocked by the Hubei CDC, until the report included market cases as well in 29/12/2019. To save face, the CCP leveraged the fact that the WCDC is right nextdoor to the market and forced official media to only say that the cases were “close to the Huanan market” but not allowing the proximity to the WCDC to be reported. https://gab.com/Flavinkins/posts/109256201942085712 All dots they moved this way (up to 1/3 of all cases) was sent to Jianghan, https://archive.md/p3K3Z https://www.researchgate.net/publication/370635299_Greater_than_the_Sum_of_its_Parts_-_Aggregated_Wuhan_COVID-19_case_data_points_to_the_wrong_side_of_the_Yangtze_River_-_Rixey_-_20230509 especially to the immediate surroundings of the market, to scapegoat it and end up causing the “unlinked cases” cluster to be closer to the market than the “linked cases” cluster, despite supposedly the linked cases should be the only source of initial human to human transmission seeding and therefore the unlinked cases should cluster near the linked cases and not the market itself. This kind of improbable-under-null-hypothesis behavior is all over Chinese “data”. And of course, Chen lived right next to the WIV BSL-4 in every dataset other than the WHO report maps—including interview datasets in the same report which where he frequents (RT-mart in Jiangxia) and which early report indicate he “lived in Wuchang” and the first hospital Jiangxia 1st Renmin hospital which he visited first and sick at the second day. Also, China WHO/WIV covered up their earlier cases intentionally—it is not plausible for 67 samples from humans taken in 01/2021 to test “all negative”. https://www.nytimes.com/2021/02/12/world/asia/china-world-health-organization-coronavirus.html They systematically moved more than 3000 cases from the lab to the market and gave “cases data” that they wanted to push for market as first outbreak site to distance from the labs. https://archive.md/rYvu3 https://archive.md/UFrSv https://archive.md/nevZy https://www.researchgate.net/publication/370635299_Greater_than_the_Sum_of_its_Parts_-_Aggregated_Wuhan_COVID-19_case_data_points_to_the_wrong_side_of_the_Yangtze_River_-_Rixey_-_20230509 Such an result of having unlinked cases closer to the market than linked cases is not expected even under the null hypothesis of market origin, which we should see unlinked cases secondary to and cluster around the linked cases, and not the market itself. https://www.researchgate.net/publication/370635299_Greater_than_the_Sum_of_its_Parts_-_Aggregated_Wuhan_COVID-19_case_data_points_to_the_wrong_side_of_the_Yangtze_River_-_Rixey_-_20230509 Not only there were an complete absence of verifiability in Chinese cases, there is direct non-circumstantial evidence that they moved up to 3000 cases from Wuchang to Huanan. In fact, it is totally not normal to have unlinked cases closer to the market than linked cases—the only way this can happen is with ascertainment bias. Only near the market gets ascertained if not directly linked to it. Base rate neglect. They did the exact same thing when claiming that all 67 “pre-Huanan checkable cases” were “serologically negative”. Again, the social media associated here say “before Jan 18, 2020”. Included all Dec cases. https://www.mdpi.com/2220-9964/9/6/402 https://ghrp.biomedcentral.com/articles/10.1186/s41256-021-00200-8 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7149375/ 135/92 cases in early peer-reviewed papers that went missing in the WHO report.






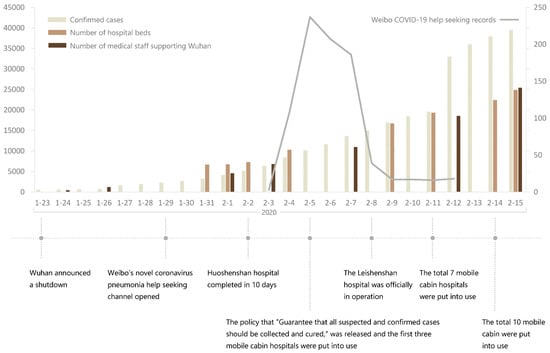
@NestCommander - Kevin W. McCairn PhD
Give this wings, it's as concise an explanation of the current state of the science as you'll get for SARS-CoV-2, PRION, Vaccines and the warfare being waged against you. https://www.youtube.com/watch?v=eDxkyX1rF_0 @CharlesRixey @BillyBostickson @still_a_nerd @Doctor_I_am_The @pizzapicklespur @Jikkyleaks @NameIsSpartacus @weve_read @BrokenTruthTV @HamEggsnSam @Parsifaler @EthicalSkeptic @ArmchairW @GasGilligan @JohnnyVedmore @dopaminergic13 @veryvirology @annunakkki @Know_More_News @RyLiberty @_HeartofGrace_ @HouseLyndseyRN @Kevin_McKernan
@NestCommander - Kevin W. McCairn PhD
https://twitter.com/r_h_ebright/status/1729164212159824154?s=46&t=wRQSWp_1VffWmS2vKQwhSA… "Former Acting Assistant Secretary of State Thomas DiNanno tells [Sky News]…that when his team unearthed explosive evidence that pointed to a laboratory leak…, the intelligence community ran interference in support of a natural origin narrative." https://x.com/daoyu15/status/1729168018763292778?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729167969534742873?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729167355098648987?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729324420622320082?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729309996591337972?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729308817584984345?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729329128690880596?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729312142879494212?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729338088911245679?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729322432891408531?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729167881777373569?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729343927344660868?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729312566239953140?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729309508219158746?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729309400148619375?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729309125472116878?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729310412578095602?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729311057519509963?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729310187012653224?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729167940287975863?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729308993607356568?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729309904702435644?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729342979910054375?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729325988176335278?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729309214131286030?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729325988176335278?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729167003418845329?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729328417479545131?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729336382592835689?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729167446370877872?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729313672324059374?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729328928719016252?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729312282541478102?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729333420160151996?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729355939214688755?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729326324379234644?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729309633842745689?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729310879529984416?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729329357955768803?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729329164577333641?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729314296969232712?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729321751727985061?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729167079838998667?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729168160191066410?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729329425798549590?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729356004482327015?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729333148692214124?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://x.com/daoyu15/status/1729324235578077646?s=46&t=wRQSWp_1VffWmS2vKQwhSA…
@R_H_Ebright - Richard H. Ebright
"Former Acting Assistant Secretary of State Thomas DiNanno tells [Sky News]…that when his team unearthed explosive evidence that pointed to a laboratory leak…, the intelligence community ran interference in support of a natural origin narrative." https://www.skynews.com.au/world-news/us-intelligence-official-linked-to-who-was-critical-in-downplaying-covid-lab-leak-theory-during-joe-bidens-90day-probe-into-virus-origins/news-story/70cec8fe1513491a421d45b12b45a8e7
@NestCommander - Kevin W. McCairn PhD
Details on the DOE Z division. Also, debunking Angie Rasmussen and ilk there again. Also None of their “data” are credible in any way.
@NestCommander - Kevin W. McCairn PhD
And no. DEFUSE does in fact propose to insert FCSes (human-specific cleavage sites in human proteins) into SARSr-CoVs. Especially when there is a mismatch to either the S1-S2 or the S2’ cleavage site, both in S2. (Such as when QTQTNSRS show up in an study-relevant Asian Sarbecovirus in 2018, where no isolated or even studied ones have an non-HT(V/A)S(L/I)LRS sequence.) If there is an infected animal, more than one spillover would happen, especially Guangdong. Why it is VERO E6 that was Wuhan growing best inside? Why all later lineages grow less effectively in it? Just fuse in a cell line that have a correct serine protease pathway. CaLu-3 also uniquely have no growth advantage to any known VOCs compared to Wuhan. Include in passage, and PRRA is more stabilized than it can ever be mutated or deleted, Proline included. arxiv.org/abs/2104.01533 An infectious clone is designed to be rescued. archive.ph/EiCQW Well, MN611520–definitely not a bat CoV. And of course, motivated reasoning like markolin can not explain how the “perfectly natural and consistent with bat sex” CoVs MN611520 and HKU4-HZAU-1 ended up one in a location without a Merbecovirus natural host (cotton but not bats or camels) and another inside an infectious clone backbone. And of course, WIV1, WIV16, Rs4874 and RsSHC014 count up to 4 published live isolates not “only 3” claimed by Shi. That is published isolates only. zenodo.org/records/570270… RaTG13 don’t grow outside immortalized kidney cells. These are just too many inconsistencies and obvious lies regarding the number of WIV Or EHA viral sequences AND isolates in their public claims. Then, there is an attrition problem where the idea that “the FCS worked impossibly well than design can anticipate” was really based on observed functions that have no bearing to pandemic potential and only recently attributed to the “specific context of the FCS”, in reality they just nee to put ENaC FCS into a QTQTNS massively mismatched S, then grow it once in VERO cells. All changes to a sequence will have half advantage and half disadvantage in the organism, but here the specific advantage of P681 “specific sequence of the FCS” is only in VERO cells, and the disadvantage however given that all natural isolate Bat Sarbecoviruses are 614D, is the complete destruction of all animal reservoirs as the incredibly unstable D614+FCS Spike got torn apart by the antibodies that would form in the animal before the FCS can emerge. The reason why no FCS exist in wild Sarbecoviruses.
@NestCommander - Kevin W. McCairn PhD
01, 03 and 04, 06 are disproven by the presence of severe and https://ayjchan.medium.com/evidence-for-a-natural-origin-of-covid-19-no-longer-dispositive-after-scientific-peer-review-af95b52499e1 https://zenodo.org/record/7169296 conclusion-disproving ascertainment bias within the CCP data. As well as cherry-picking of early genome data. https://www.researchgate.net/publication/362806429_Unwarranted_exclusion_of_intermediate_lineage_AB_SARS-CoV-2_genomes_is_inconsistent_with_the_two_spillover_hypothesis_of_the_origin_of_COVID-19 https://gab.com/Flavinkins/posts/109152915624650793 08 is disproven by the fact that there were no by-month data for Xiao Xiao et al, and there being no evidence of December 2019 animal sales that can be proven through an image that contained either features or metadata that permit them to be dated. 05 is simply wrong. Yunnan is the hotspot, Guangdong is linked to wildlife trade. Wuhan is neither. 09 is disproven as the specific pattern of RE sites are not directly linked to spillover probability and the probability that it is easy to clone with the standard BsaI/BsmBI through natural recombination is less than 1/32. https://gab.com/Flavinkins/posts/109255356915252021 03 is disproven since despite all the efforts, FCS continue to elude all efforts to find it. https://archive.ph/k7S6T https://archive.ph/Ga1iI https://archive.ph/vUy8n 07 is disproven by Marburg virus and also RSV. Especially RSV, which originated in polio vaccine research. https://gab.com/Flavinkins/posts/108661483685033341 https://www.bmj.com/content/344/bmj.e2398/rr/599724 As for 10, the actual graph seen—show humans and largemouse bass not raccoon dogs. Humans and several livestock species in the “志翔冻品商行” near the toilets are the only species with any mutual information at all, highly consistent with boot and suit contamination. All metric positive correlation is observed only in humans and the entirely non-susceptible livestock species sold near the toilets. Worobey failed to address the fact that “the neighborhood of Huanan market” was used during the early case collection process—he opted to remove directly linked cases but none of the critical annex D5 cases that were collected from “the neighborhood of Huanan market”. Exactly 32 of these dots exists, = 59-27. https://gab.com/Flavinkins/posts/109228312723838390 https://gab.com/Flavinkins/posts/109443089504009640 https://gab.com/Flavinkins/posts/109148677382700486 https://gab.com/Flavinkins/posts/108825678909519515 These were cases that were collected because of their geographical proximity—collect if lived in the neighborhood, regardless of hospital. Collect from other hospitals only if exposed to market. Collect from hospitals near the market. https://gab.com/Flavinkins/posts/109386394452941367 https://gab.com/Flavinkins/posts/108830214433800007 https://gab.com/Flavinkins/posts/109169722840473497









@NestCommander - Kevin W. McCairn PhD
https://gab.com/Flavinkins/posts/109205261283826972 ReCCA is tautological and fictitious. https://gab.com/Flavinkins/posts/109863181504837302 https://gab.com/Flavinkins/posts/109465063042828622 https://gab.com/Flavinkins/posts/109255356915252021 BtSY2 is sequenced in 2018. https://gab.com/Flavinkins/posts/109399710986742685 BANAL is in the hands of the DOD in 2017. https://gab.com/Flavinkins/posts/109340247585238829 https://gab.com/Flavinkins/posts/109800300869616862 The only thing in the market that meaningfully focus the abundance of SARS-CoV-2 is “closest to the toilets”. As all non-human “susceptible species” failed to correlate positively with SARS-CoV-2 consistently or with significant mutual information. With no actual evidence of natural infection in any of the so-called “susceptible species” at all. Contamination artifacts from samplers, and not actual animals, Or even vendors, created all of the “positive environmental samples” in the market. A fact which fraudulently bleaching the toilets before sampling can not hide, And which they failed to realize or create the required secondary spillover outbreaks in other locations Which all zoonoses have.





@NestCommander - Kevin W. McCairn PhD
@NestCommander - Kevin W. McCairn PhD
Except that 1: ZC45 and HKU3 are not SARS-CoV-1 or SARS-CoV-2. And 2: the raccoon dogs are locally wild-caught in Wuhan, tested and negative and 3: the animal-specific viruses are in perfect positive correlation with the animals, the SARS-CoV-2 is in consistent correlation with significant mutual information only with Homo Sapiens. And 4: the FCS itself optimized to cell cultures, HAE/VERO and CaLu-3, mutated in live hosts.
@NestCommander - Kevin W. McCairn PhD
Because Homo Sapiens is still the only species that they can get infected at all, if you zoom in and correlate between animals and viruses, You get animal-specific viruses being correlated strongly positively to the animals, and SARS-CoV-2 being positively correlated consistently or with significant mutual information only with Homo Sapiens.
@NestCommander - Kevin W. McCairn PhD
archive.md/yyX0Z archive.md/iw1Pz archive.md/4rVph archive.md/DChUL None of the “susceptible species” found in the market had a single report of natural infections by SARS-CoV-2, anywhere in the world. Closest and second closest stall to the toilets=👢👖🥼contamination have the highest frequency of happening among sampling. Neither civets nor raccoon dogs are susceptible at all to natural infections with SARS-CoV-2 with zero reported cases anywhere in the world. None of the “susceptible species” found in those “wildlife stalls” have been reported anywhere in the world neither China nor Europe Japan Vietnam of an natural infection by either SARS-CoV-2 or even a relative of it. Nor bamboo rat, hedgehogs or porcupines. No Guangdong spillovers directly rule out infected animals in the wildlife trade—specific supply from Yunnan to Wuhan can not support an economically viable farm because the extremely low consumption rates in Wuhan. archive.md/e3615 archive.md/vWjZl Homo Sapiens is the only species that remain consistent and significant positive correlationship within those “wildlife stalls”. https://archive.md/gvHfw https://archive.md/LJzSO https://archive.md/4cCHG https://archive.md/gkquN And the real determinant and the primary confounding factor for “which stall have the most positive samples out of all samples https://archive.md/JSQvc https://archive.md/csYBM Is “closest to the toilets”, also “closest to the entrance to the market” for the wildlife stalls. https://archive.md/vlAgp https://archive.md/mwT8i This is because all of the positive results from the “stalls” were caused by contamination caused by the samplers either smeared out from the toilets or brought in from the outside. This is also the reason why nearly all of them were found below step height, the height below which accidental trampling and kicking are highly likely. https://archive.md/FskYn https://archive.md/lI04H All that were left were either directly from the PPE of the samplers themselves https://archive.md/rj1pV https://archive.md/VNr75 Or are located right where contamination either on the surface (near walls or doors to be rubbed against, give PCR+) or on the sample tubes (awkward, crammed location making it difficult to not touch the lip of the sample tube onto sampler suits, give PCR-) are likely. These locations are also the locations that are most likely being sprayed (below step height) and the max normal operational height (below waist height for devices recorded on photo) by a sprayer or other dispensing device equipped by a “hazmat suited worker” that entered from the main entrance and was seen fiddling around with the environmental surfaces in 02/01/2020–without collecting a single environmental sample despite operating equipments in a way that look like virology work from afar. As for any direct vendor-handled surfaces, the RNAse 7 activity (skin defense ribonuclease, attack membrane-bound pathogens especially with an incomplete glycan shield) inside these samples are so strong that all SARS-CoV-2 RNA is completely destroyed within a single day between collection to sample processing. https://archive.md/2PM9Y https://archive.md/RirQ7 https://archive.md/NeybM https://archive.md/BWZJL https://archive.md/CTP3i https://archive.md/ETjzS The possibility of the same infected sampler that dropped the contaminated PPE in 31/12/2019 then contaminated all of the wildlife stall samples whenever he/she enters through the toilet area for that sample run, can not be discounted as none of these subsequent runs contained a lineage read.
@NestCommander - Kevin W. McCairn PhD
They are also compatible with a scenario that the same culture that was used in the initial planting work was eventually used to adulterate A20 when demanded by the CCP after learning about the lineage A issue. https://archive.md/LJzSO https://archive.md/4cCHG The first set of samples https://archive.md/gkquN contained human residues inside that became the only species with consistent positive correlation or one with significant mutual information with the SARS-CoV-2 reads in the market, then with the demand to blame snakes and to make sure humans don’t make its way into the samples, they stained the mid-phase samples with obvious amplicon artifacts and “tested” the rest with the highly cross-reactive PREDICT ORF1ab primers to manufacture “positivity” which none contained a real read of SARS-CoV-2. Finally The very last positive wildlife stall samples https://archive.md/13bdP, taken after the cleaning of the toilet area https://archive.md/rSaO9 , contained no mammalian species other than Homo Sapiens, which represents the “pure” form of the contamination within the wildlife stalls that is brought into the market by a sampler. They now use a mixture of animals to add to their to-be-tampered-with sample, derived from an assemblage of frozen products taken from the market so that they can say that the positive human/virus correlation, persisting even into these samples, are caused by “differential preservation”—Unfortunately the animal mix did not contain any mammals, meaning that they in stead become direct evidence that lab based cultures of SARS-CoV-2 (infections and culture supernatants give mainly the virions and a genomic profile for the viral RNA, culture lysates gives the intracellular transcriptomic profile for the viral RNA and host Mitochondrial DNA that is seen in these final “storehouse” samples.)
@NestCommander - Kevin W. McCairn PhD
https://web.archive.org/web/20231213033617/https://en.rattibha.com/thread/1714233859276149199 https://archive.md/73xfX Once inside “wildlife stall A”, The only correlation crashed to Homo Sapiens. Others crashed to zero one metric or another. https://archive.md/8nN3k Same as in the positive samples. Ask “which species shed the SARS-CoV-2 where it is found” yield “Homo sapiens”. The animals correlated with their native viruses, and not SARS-CoV-2. https://archive.md/BrQy7 https://archive.md/eoaMn
@NestCommander - Kevin W. McCairn PhD
The “all samples” merely asks “which species is sold most uniquely in the stall closest to the toilets” with much of the animals being on the ground, entirely wrong sections, https://archive.md/eoaMn and a heavily confounded distribution pattern centered around the toilets same as W4-26-28 in Jan 01. https://archive.md/FskYn https://archive.md/gvHfw Also, Raccoon dogs are in fact, never found infected in nature at all, neither China nor Europe. archive.md/g2L31 archive.md/OIHeo Sampler contamination and cross-contamination. archive.md/LJzSO archive.md/VNr75 Never an infected vendor or animal. In fact, the samples in the market follows the rule which a positive sample archive.md/CTP3i archive.md/ETjzS archive.md/BWZJL must be contacted by samplers.🥼👖👢=positive. archive.md/NeybM archive.md/2PM9Y archive.md/RirQ7 And not frequently handled by vendors.🥩🥬🍄🛁📻☎️📦🧺=negative. Another oddity, is that for the entirety of the market, none of the personal items of vendors and none of the frequently directly handled objects from boxes to baskets to cashiers, were positive. This is because SARS-CoV-2 RNA specifically is extremely unstable when on surfaces that are highly touched by a human—D614 especially because the Spike were too sparse to form a shield defending against invading RNAse 7. archive.md/LJzSO In fact, all of the positive samples in that “wildlife stall A” and the majority of the positive samples in the market is below step height—they are contaminated by the toilets and archive.md/4cCHG archive.md/FskYn is the reason why the stall with the most positive samples out of all samples is the stall closest to the toilets. In both Jan 01 and Jan 12. The superfluous nature of SARS-CoV-2 is therefore evident—not shielded inside solid tissues because all of it is shed by humans, and with an incomplete glycan shield, any that landed where human skin contact frequently (vendor items like cashiers, knives and chopping boards, baskets, boxes, water cup = least stable like bangladesh banknotes, not frequently touched locations like bases of machines or non-door-knob locations of cages, loading surfaces of grounds, scales or carts = more stable like sewage. Sampler PPE like gloves or shoe covers=most stable, and the main vectors in spread contamination to the objects and sample tubes.) got destroyed by RNAse 7. No positive above waist height, because these locations are where archive.md/FskYn samplers can exercise more care and it is less likely especially for untrained private contractors or volunteers briefed only once to avoid contaminating (enough room to avoid touching the surface or sample tubes, high enough so samplers can see to avoid rubbing with their suits. Below waist height on the other hand, not even professionals can avoid kicking or trampling due to lack of line of sight, and rubbing of the suits onto the surfaces are inevitable. Crammed locations where samplers need to support their own weight with their hands, breaks even the most professional of techniques and lead to high chance of sample tubes lips being contaminated.). 👢on 🛒🏔👣=PCR+/NGS+, 🥼on🧪=PCR-/NGS+ aligning over the primers and negative before and after. archive.md/tlfNr archive.md/GvRcD https://assets.researchsquare.com/files/rs-885194/v1/78e0e6ce-4a76-48de-9f5c-76bab452bbe6.pdf?c=1665607885
@NestCommander - Kevin W. McCairn PhD
The effect of spreading contamination out of the toilets and the main entrance of the market is evident not only in that both Jan 01 and Jan 12 have the stall with most positive samples out of all samples being the stall closest to the toilets, but also evident in the form of multiple samples from nearby stalls with only human DNA, no human cases and positive samples. They represent the purest form of sampler-linked contamination. https://archive.md/gvHfw The only thing governing the probability for positivity of the environmental samples is “closest to the toilets” and “closest to the main entrance of the market”. And bleaching it before sampling https://archive.md/rSaO9, just archive.md/13bdP lead to samples with only human DNA and no other mammals at all in the last samples hyper specifically taken from the “wildlife stalls”. All they managed to get in an effort to verify the ORF1ab primer-overlapping and Jan01-negative/PCR-negative contaminated sample tube are amplicon artifacts, false positives without reads, cultures of the virus from in-lab contamination and not a single positive of any kind on the site for Q37 any more.
@NestCommander - Kevin W. McCairn PhD
https://assets.researchsquare.com/files/rs-885194/v1/78e0e6ce-4a76-48de-9f5c-76bab452bbe6.pdf?c=1665607885 Unlike animals including livestock, humans are neither sold nor butchered at the market. Their CoVs degraded catastrophically after 01/01/2020 and completely after 12/01/2020 leaving only artifacts behind. archive.md/13bdP archive.md/FskYn archive.md/gvHfw archive.md/4cCHG Animals that are sold and butchered at the market have their CoVs remain stable and are the only CoVs left detectable in February 2020. Bane of the Zoonati: W4-26-28. Especially W4-28 which don’t have human cases inside. Only 1 out of the 5 samples have any wildlife reads at all. Closest to the toilets. Trample contamination clearly evident. Guess which species never have a single sample which it is not present? Homo Sapiens. In both Jan 01 and Jan 12 the stall with most positive samples out of all samples are the stall that is closest to the toilets in the market. Guess why. Guess why they never hint on Pearson correlation or the positive samples at all? The formulation of the problem “which species shed the SARS-CoV-2” gives an easy verification for species correlation by looking in samples where you actually see the SARS-CoV-2. This process also normalizes out any spatial confounding factors especially when confounded by pathological spatial distribution of the samples (all PCR+ samples in one stall closest to the toilets), where “which species is sold uniquely in a stall” guarantee by lottery fallacy a random species (each stall have one to two species that are uniquely sold inside and have low prevalence in the overall market), but actually entering the stall or the samples where you see the SARS-CoV-2 result in a collapse of correlation where no species except Homo Sapiens landed on the same sections of the ground as SARS-CoV-2. And of course, despite also being enteric like animal CoVs, SARS-CoV-2 failed to persist in the market, indicating it being not inside solid tissues like the animal CoVs, and therefore, not able to withstand either RNAse 7 degredation or cleaning agent application like animal CoVs could. Despite evident strong fecal and rectal shedding, persistence was not guaranteed for SARS-CoV-2 because it is not found in butchered animal tissue, which have safely and durably protected all animal viruses in the market all the way down to the end of February 2020. In addition to the observation where nearly all positive samples are found below step height and none of them being found above waist height, where incontrovertible proof of boot and suit contamination exist in the form of positive samples with neither human cases nor wildlife DNA, (which even followed the expected pattern as boots first bring the virus inside from the main entrance, then get additionally contaminated by wildlife DNA from the W6 junction) The stark contrast between highly persistent animal CoVs and SARS-CoV-2 which degraded to nothing but artifacts within mere 12 days, clearly indicate that the SARS-CoV-2 in the market is not shed by animals.
@NestCommander - Kevin W. McCairn PhD
Because SARS-CoV-2 is not found in any animals which would be butchered and their tissues and guts and noses split open and spilled onto the market surfaces, the human respiratory and enteric shedding that deposited the SARS-CoV-2 into the toilets archive.md/gvHfw and then smeared onto the market stalls, can not shield the virus from being destroyed either by RNAse 7 activty or cleaning of the market. archive.md/LJzSO archive.md/4cCHG Consequently, only SARS-CoV-2 degraded over time in the market, nearly completely degraded in Jan 12 and completely degraded afterward leaving nothing other than amplicon artifacts and obviously in-lab cell culture lysate adulteration with no susceptible animals inside and with the intracellular human mitochondrial and viral Transcriptomes still intact archive.md/13bdP (they decay to only MtDNA and viral gRNA within two days ex-vivo unless in an -80C lab freezer—environmental surfaces like the market or even significant storage time after leaving the growth environment in working conditions can definitively not provide such conditions e.g. all the samples before them). archive.md/YGDiK Animal CoVs on the orher hand, persisted, with reads consistent with real, artifact-free genomes, and with no significant concentration changes that can indixate degredation, in Jan 01, Jan 12, Jan 26-27 Drains (the only SARS-CoV-2 read at that point was found in a sewage well on the opposite end as the wildlife stall that is connected to the municipal sewage system but not the wildlife stalls) archive.md/8nN3k and all the samples afterward, including samples taken after 15/02/2020. No indication of significant degradation or quality change of animal viruses and animal CoVs are observed in any sampling date indicating that they last at least 3 months and most likely longer, unlike human-sourced SARS-CoV-2 that last no longer than 15 days without sterile conditions. And unfortunately bleaching the toilets https://archive.md/rSaO9 https://archive.md/nAqKp even amplifies this problem—wildlife CoVs persisted, and no SARS-CoV-2 except adulterated using an laboratory culture so fresh that even the human Mt transcriptome (don’t last in the environment for 2 days or longer) and SARS-CoV-2 intracellular transcriptome (don’t last in the environment for a week or longer) are left inside.
@NestCommander - Kevin W. McCairn PhD
Unfortunately, all that existed for Q61 and Q70 are the result of cross-contamination from Q64 and Q68/Q69, All of which are on the ground and archive.md/YGDiK are the result of either lower level boot and foot contamination x.com/daoyu15/status… x.com/daoyu15/status… Same as Q64/Q68/Q69 (stepped on>kicked for contamination). x.com/daoyu15/status… All that archive.md/73xfX archive.md/8nN3k archive.md/FskYn archive.md/gvHfw exist for Q37 is the contamination of a sample tube by the gloves and suits of the samplers. The swab is clean, PCR-. The tube lip is contaminated, NGS+ with alignment over the CCDC SARS-CoV-2 ORF1ab primer pair. archive.md/LJzSO archive.md/4cCHG PCR+/NGS+ mean the virus is present in the location. PCR+/NGS- or NGS(artifacts) mean you are using the incorrect primers (all incidents happened with the PREDICT ORF1ab only primers). archive.md/rj1pV PCR-/NGS+, especially when archive.md/csYBM the primers are aligned over by NGS reads, indicate that the samples have been catastrophically contaminated as NGS is a more complicated process that are far more prone to contamination compared to PCR. archive.md/13bdP The stall for Q37 is negative at Jan 01. They then went on sampling the same stall including the “freezer” twice afterward, attempting to verify the “sample” they considered most promising. Bringing in artifacts elsewhere and samples without a read (the only sample with a real SARS-CoV-2 read at all gathered using the PREDICT ORF1ab only primer pair was a sewage well connected to the municipal sewage system on the exact opposite to the “wildlife corner”.), but never SARS-CoV-2 reads any more. This fact is also reinforced with the intriguing observation where Q37 is found to be in the same correlational series between SARS-CoV-2 and Homo Sapiens as other Q* samples. Attempts at sampling the archive.md/FskYn archive.md/gvHfw archive.md/4cCHG archive.md/csYBM archive.md/rj1pV “storehouse” just ended up with a total catastrophe—archive.md/13bdP the sampling team brought in in-lab culture contaminants, not even aged for more than a day, into the sampling sites again when they suited up in their lab and entered the location. Impossibly fresh intracellular Homo Sapiens and SARS-CoV-2 transcriptomes, neither capable of lasting for more than two days ex-vivo in that condition, ended up contaminating the samples and without a single read of a susceptible animal inside those “samples”. Sampler contamination and cross-contamination. archive.md/LJzSO archive.md/VNr75 Never an infected vendor or animal. In fact, the samples in the market follows the rule which a positive sample archive.md/CTP3i archive.md/ETjzS archive.md/BWZJL must be contacted by samplers.🥼👖👢=positive. archive.md/NeybM archive.md/2PM9Y archive.md/RirQ7 And not frequently handled by vendors.🥩🥬🍄🛁📻☎️📦🧺=negative. archive.md/HlJ9o archive.md/nAqKp archive.md/rSaO9 They also put bleach onto the toilets and the mahjong room before sampling them. This is a clear move to cover up. archive.md/csYBM And no the “stall” W5-NA was sampled on the inside 27/01/2020, negative. (Not even animal CoVs were there) The toilets is the real contamination source. archive.md/C5oal archive.md/RSsS7 and yes only Homo Sapiens positively correlated with SARS-CoV-2 consistently in all metrics, or formed any kind of line or grouping pattern at all that allow the abundance of one to be estimated at above-random success rate and precision using the other (e.g. have any significant mutual information with SARS-CoV-2). archive.md/0O2TN archive.md/GjlEx
@NestCommander - Kevin W. McCairn PhD
If you examine the inside of “wildlife stall A”, then all you see is boot prints and suit marks. None of it is animals. All metrics now favor Homo Sapiens as the most likely source of the SARS-CoV-2 sequences there. The wildlife stalls all sold susceptible animals. They only sampled wildlife stalls in Jan 12. Then positives are found closest to the toilets because that is where contaminated suits and boots most likely rub trample and kick. No different from W4-28 and W4-26-28, really. Not forming a line on the correlation diagram=no mutual information=spurious. That is why it dissolved completely when asking “which species shed the SARS-CoV-2” in slices where the analyte concentrations aren’t 0. That is also why they fraudulently bleached the toilets before sampling them.
@NestCommander - Kevin W. McCairn PhD
And this is how you ID a spurious result. If the correlational diagram show a neat line indicating an consistent increase in the count of one candidate factor as the target analyte increased, it indicate that the target analyte is probably caused by the candidate factor with a consistently raising minimum of the factor per analyte suggesting that all of it is brought in alongside this candidate. If the correlation diagram show an randomized pattern or even a line of negative slope, especially when nearly all of the target analyte is found in one place, then it is probably that it is just the one place have one or few potential candidates that are less abundant elsewhere, which with the 25+ candidates in the market sample correlation analysis guarantee 1-2 for every stall (and most of which are on the ground just like anything trampled from the toilets, with entirely different reasons). If you can not use the concentration of the target analyte to reliably predict the concentration of the candidate, or come up with an result that the more target analyte there is the less candidate there is where the target is found, e.g. an absence of or negative mutual information, then it is most likely spurious and extremely unlikely that candidate yielded or is brought alongside the target analyte. Causation are bijective. Confounders are injective. Spurious correlations are correlated only in some metrics and slices but not all. Inconsistency between different slices and metrics indicate an lack of true causation and likely confounder that makes false positive in some but negative in the other. A consistent positive correlation in almost all metrics and no negative correlation in any metric, Like Homo Sapiens, indicate that there is true causation that some disruption may have occurred. Species that have “positive” correlation only in some metrics out of a single slice, (not even all the slices examined for that date), but negative or zero in all the other metrics and slices with the mutual information metric yielding negative and zero only regardless of slice, like oriental rat snake or malayan porcupine, are spatially confounded—they are “the most unique species found in the 1 stall at that slice that was closest to the toilets”, and since every stall have one such species, they represent false discovery by lottery fallacy, and fails when any other slices are used. They also failed to form a line on the correlation plot which indicate that there is no causation and the animal did not shed the virus where it was found.
@NestCommander - Kevin W. McCairn PhD
archive.md/csYBM And no the “stall” W5-NA was sampled on the inside 27/01/2020, negative. (Not even animal CoVs were there) The toilets is the real contamination source. archive.md/C5oal archive.md/RSsS7 and yes only Homo Sapiens positively correlated with SARS-CoV-2 consistently in all metrics, or formed any kind of line or grouping pattern at all that allow the abundance of one to be estimated at above-random success rate and precision using the other (e.g. have any significant mutual information with SARS-CoV-2). archive.md/0O2TN archive.md/GjlEx This result is consistent with the theil-sen estimator, which explicitly measures mutual information through the use of all slopes in the data points, which measures the predictive power of the other data points and one metric of one data points to the other metric of the data point. You can easily distinguish between common tertiary cause (confounded) from true causation by looking with increasingly finer grain of resolution, especially where the data points aren’t 0. Unlike spurious correlations from Confounding factors which ends at the resolution where the factor acts on, True causation stay correlated in every resolution and in any set of data points especially where the data values aren’t 0. In fact, confounding factors often crash in correlation quite early before that. “Do pirates get less prevalent the hotter it is when normalized agains temporal trends, where many things have correlation with time”? Is the same as “Do SARS-CoV-2 correlate with animals at all when normalized against confounding pathological distribution of insufficient spatial spread, which each stall including one closest to the toilets have several separate unique species?
@NestCommander - Kevin W. McCairn PhD
And it really only need “VERO cells and HAE cultures” to adapt to this.
@NestCommander - Kevin W. McCairn PhD
What those analyzers think: “many different precise and specific pieces recombine into SARS-CoV-2”. Reality: an unpublished but readily sampled SARS-CoV-2 progenitor spread fragments over time into multiple locations. Some end up in the published samples. Assumption of recombinant origin is almost always because of a lack of published samples sufficiently close, especially with multiple of samples of comparable distance. One specific contig is fished out of the assembly graph from a bucket of reads on the criterion that it meet the RE site requirement, before being used for cloning. Natural recombination will not pick that specific contig over any other possible assembly graphs for spillover. What they claim: “Only QTQTNS + PRRA would work!” Reality: they test an appropriate human FCS on all manner of Spikes, especially when they see an S1-S2 site “cleavage sites in S2” (that are structurally homologous to 757/900 for HKU1 “other coronaviruses”) with the example of “667/792 for SARS” that have deviated from the established consensus of spillover-capable SARSr-CoVs up to the point if DEFUSE. And it just happened that the closest human FCS to the first mismatched S1-S2 they see is the hENaC FCS, introduced by adding “ARRA” to “RSVAS”. Several versions tested and one ended up working. https://gab.com/Flavinkins/posts/109205261283826972 ReCCA is tautological and fictitious. https://gab.com/Flavinkins/posts/109863181504837302 https://gab.com/Flavinkins/posts/109465063042828622 https://gab.com/Flavinkins/posts/109255356915252021 BtSY2 is sequenced in 2018. https://gab.com/Flavinkins/posts/109399710986742685 BANAL is in the hands of the DOD in 2017. https://gab.com/Flavinkins/posts/109340247585238829 https://gab.com/Flavinkins/posts/109800300869616862




@NestCommander - Kevin W. McCairn PhD
It is not just that SARS-CoV-2 Wuhan grows best in VERO cells out of all variants.
@NestCommander - Kevin W. McCairn PhD
Some earliest patients harbored inside their QS specific S1-S2 deletions that can form only in VERO E6. https://gab.com/Flavinkins/posts/109640519028841414

@NestCommander - Kevin W. McCairn PhD
archive.md/HlJ9o https://archive.md/rSaO9 https://archive.md/13bdP https://archive.md/nAqKp They also put bleach onto the toilets and the mahjong room before sampling them. This is a clear move to cover up. https://archive.md/rj1pV https://archive.md/FskYn archive.md/csYBM And no the “stall” W5-NA was sampled on the inside 27/01/2020, negative. The toilets is the real contamination source. https://archive.md/LJzSO https://archive.md/4cCHG archive.md/C5oal archive.md/RSsS7 and yes only Homo Sapiens positively correlated with SARS-CoV-2 consistently in all metrics, or formed any kind of line or grouping pattern at all that allow the abundance of one to be estimated at above-random success rate and precision using the other (e.g. have any significant mutual information with SARS-CoV-2). archive.md/0O2TN archive.md/GjlEx What they tried to hide with this: The fact that “closest to the toilets” is the only factor that governs where you are going to see positive samples the most in the market—there is no difference between W4-28 and W4-26-28 in 01/01/2020 and W6-29-33 in 12/01/2020 in term of where the virus came from and why they have the highest positive sample count out of all sample counts in their respective sampling runs. Sampler contamination and cross-contamination. archive.md/LJzSO archive.md/VNr75 Never an infected vendor or animal. In fact, the samples in the market follows the rule which a positive sample archive.md/CTP3i archive.md/ETjzS archive.md/BWZJL must be contacted by samplers.🥼👖👢=positive. archive.md/NeybM archive.md/2PM9Y archive.md/RirQ7 And not frequently handled by vendors.🥩🥬🍄🛁📻☎️📦🧺=negative.
@NestCommander - Kevin W. McCairn PhD
Unfortunately, none of the “drain” samples contained actual SARS-CoV-2 reads. They have animal CoVs, demonstrating that the SARS-CoV-2 in the market isn’t present in animal tissues and does not last past 12/01/2020 as legitimate reads, and that are also happens to be exceptionally cross-reactive to the ORF1ab only tests used.
@NestCommander - Kevin W. McCairn PhD
Cold hard reality: there is nothing special to W6-29-33 compared to W4-26-28. Closest to the toilets is what that govern where the virus would be found. Fact: w6-29-33 and the market supply chain did not get shut down at all. The former operated as “金秀山家禽批发商行” up to the end of 2021 and the latter operate still to supply other markets in Hubei, archive.md/yyX0Z archive.md/iw1Pz archive.md/4rVph archive.md/DChUL evidenced by the fact that these suppliers including the exact animals sold were readily sampled in Jan-feb 2020. This included also specific sampling of several species inside the stall itself, also negative. Their animals tested negative and no outbreaks happened. They represent the full inventory of raccoon dogs and siberian weasels in the market. The Hubei supply farms then represent the full inventory of many other species. The only Yunnan supplied animals all landed on the wrong stalls, testing negative even when sampled as is in the market. archive.md/e3615 archive.md/vWjZl
@NestCommander - Kevin W. McCairn PhD
This is the reality: VERO cell adaptation is max in Wuhan compared to bat or VOCs.




@NestCommander - Kevin W. McCairn PhD
And this picture is worth a thousand words—“VERO cells and HAE cultures” adapt the virus like this exactly.




@NestCommander - Kevin W. McCairn PhD
And also fact: you really can not 1: ignore the 50+ strains of bat SARSr-CoVs identified by the EHA, still not published—anything sufficiently close won’t be “mosaic recombinant”. And 2: the China claim that there were no cases before the market got completely self-debunked when they then went on saying that 67 samples in an 2021 Wuhan of >4% seroprevalence “tested all negative”. Numerous anomalies leaked in regard to early detections that is incompatible with a market origin. Sinterklaas/Faucier: Wei Jingsheng: WHU student reports: First public Wechat Spike in “SARS”: Wuhan lab banned from being mentioned the first official declaration(nobody is allowed to doubt the constructed official narrative): Callahan: Murdered Zhou Yusen with body bags found outside the WIV: It is impossible to have 67 wuhan residents sampled in 2021 to test all negative for SARS-CoV-2 antibodies. Blatant ben HU lie on “not working with SARSr-CoVs” Still operating illegal labs in the U.S. They screwed up with the military games it seems. And it is not unusual to see samples in China being sequenced only months to years after initial collection and culturing. Hong kong suspend Wuhan mail over Nov-Dec 2019. https://gab.com/Flavinkins/posts/108890415550666278 Chinese communities in Italy show evidence of prior immunity. https://gab.com/Flavinkins/posts/108952233844548674 Mean time of earlier SARS-CoV-2 related discoveries=mid November 2019. Clinics—even in mid December 2019 in central Wuhan—got 10 times more cases than usual in mid November and only worsens through December and January. https://archive.md/VXtu9 Cases begun to take off and it have reached “more than 600 a day” just before the start of 2020 that they completed all of their preparation job and got all their PPE they can get before 01/01/2020. Then ascertained cases alone flooded the hospital just about 10/01/2020. https://gab.com/Flavinkins/posts/109248812361151175 https://archive.md/VXtu9 Here is evidence that widespread silent outbreak have already happened inside Wuhan before it reached the HSM, with the Tongji and Union hospitals already seeing novel coronavirus inside the Wuhan CBD in 6-8 Dec 2019. Of course this is again never official. Why trust only the official cases data with a known bias and severe censorship? https://archive.md/UIBkB







@NestCommander - Kevin W. McCairn PhD
The only thing governing the probability for positivity of the environmental samples is “closest to the toilets” and “closest to the main entrance of the market”. archive.md/yyX0Z archive.md/iw1Pz archive.md/4rVph archive.md/DChUL Also here is a result on the raccoon dogs and the inability for the species to become infected in nature. archive.md/n9o0f All non-human mammals archive.md/7doR8 archive.md/0A24q at most landed on different sections of the ground and correlation fails upon entry to that “raccoon dog stall”. archive.md/Ttn5P archive.md/JSQvc Coincidence caused by pathological spatial distribution on the most uniquely found species in the stall closest to the toilets archive.md/gvHfw have high R^2–all landed on different sections of the ground and fails upon entry into the stall. archive.md/0A24q True causation remain positively correlated when looking at the positive samples or when you enter the site of the pathological spatial distribution. archive.md/csYBM Also, in order to test positive in Gao et al, a sample archive.md/CTP3i archive.md/ETjzS archive.md/BWZJL must be contacted by a sampler. 🥼👖👢=positive. archive.md/NeybM archive.md/2PM9Y archive.md/RirQ7 Must not be frequently handled by a vendor. 🥩🥬🍄🛁📻☎️📦🧺=negative. There is a reason why the theil-sen correlation, a quantifier of mutual information, show Homo Sapiens as max correlated wherever any species in the “susceptible mammals” category (wildlife and humans) show correlation at all.
@NestCommander - Kevin W. McCairn PhD
And of course, Polymerase stuttering is exclusive to Influenza and other negative ssRNA/DNA viruses (requires an “unzipping last” transcription mode which the nascent transcript is unbound from the template immediately after synthesis) and in fact, the S1-S2 is a cold spot of recombination because CoV template switching depends on TRS-B binding to Nsp7 and not by “stem loops”.

@NestCommander - Kevin W. McCairn PhD
Reverse transcriptases are critical in understanding the dynamics of SARS-CoV-2 genomic insert evolution. “How many billion hosts are needed for the GISAID sequences to appear? (2 billion) what kind of methodology they were sequenced with (because multiplex PCR+bad primer batch=additional sequences end up being inside the amplicons especially same supplier of primers used), and how many hosts a wildlife farm is able to harbor at maximum? (Less than 2 or other markets that the farms have to supply to because of the low animal counts in Huanan would be infected and have primary outbreaks) What happened to those “lineages”? (They disappeared upon the exhaustion of the specific primer batch they were sequenced from, and never continue circulation within sequences for more than ~2 weeks)” . It is something that they can not answer. no proline no VERO growth Destroyed in VOC evolution and no glycans to be seen https://gab.com/Flavinkins/posts/111398506038803573 The proline is required for efficient growth in VERO cell cultures and all live hosts despise it. With nearly a billion sequences available now all manner of sequencing error and biomaterial manufacture errors can happen. That is why none of the pandemic era inserts in covid lasted for more than the length of time which a batch of primers ordered from a company usually last in the testing and sequencing labs. If you look for dirt this big a base number and the high random and systematic error rates would guarantee something you will see. Also the “potentially real inserts” are all from within the SARS-CoV-2 genome. No CGG-CGG pair anywhere within the genomes of the closest relatives of SARS-CoV-2.

@NestCommander - Kevin W. McCairn PhD
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7239183/ All 3 Lines of PO argument have been debunked. There is no evidence of O-linked glycans in the actual SARS-CoV-2 spike for cells that are relevant to real live hosts or immune systems. https://zenodo.org/record/6849652#.ZKUlyCV6slT And RmYn02 having 2AA shorter than normal not 3AA longer. vixra.org/abs/2010.0164 All RmYN02 “proves” is the motivated reasoning of the final PO public arguments. Even the authors themselves do not believe their arguments point strongly toward anything.
@NestCommander - Kevin W. McCairn PhD
And of which all that existed were just viruses that were smeared out of the toilets and shed by researchers, and which nearly all positive samples are below step height, no positive samples are above waist height and that none of the highly touched locations are positive. Humans=shed in the toilets and feces are stuck all over the boots and suits and shoes and clothes of the samplers and vendors alike. Suit-stained walls doors and legs of desks (but not tops of tables), boot-kicked machines, cages, carts, scales and of course the ground itself which is the dominant sample type for positive samples. And suit-stained sample tubes where the swab is clean but the lip isn’t (causing PCR-/NGS+). In fact all animals that can be infected at all shed in their feces for SARS-CoV-2 RNA. Yes. SARS-CoV-2 have enteric tropism and shed RNA in feces for both animals and humans. You know that transfer contamination is the dominant if not the only mechanism for market environmental samples when there are also samples that are +ve in both PCR and NGS, but linked neither to human cases nor to wild animals. Even the presence of materials from different origin within the samples are consistent with transfer contamination with a pathway that first go through the toilets and then go through the W6 junction, getting SARS-CoV-2 on the former and wildlife material (on only a fraction of the boots) on the latter, independently. More samples with neither cases nor wildlife DNA are found south of the W6 junction than north of it, but such samples also exist north of the W6 junction. This is consistent with the virus being brought in from the entrance/toilets, contaminating stalls where there is also a focus to stalls with human cases. When boots stepped through the W6 junction, some of the boots also have wildlife DNA stuck to them, bringing it alongside when sites north of the W6 junctions were kicked or trampled. but not all of them were and there exist also incontrovertible proof of samples with neither human cases nor wildlife DNA found also here. Good and specific PCR primers, like Jan 01/Jan12 ORF1ab+N, and you should have PCR+ before NGS+. Bad and cross-reactive PCR primers like an ORF1ab only primer, and you are going to have PCR+ anytime you see material from the same family you are trying to test on (Embecoviruses cross reacted with their ORF1ab primers—and these animal CoVs are the only real grounded CoV consistent with samples of the expected age at sequencing found here in the specified time). However, PCR-/NGS+ is something that should never happen nomatter which primer pair you use (cross-reactive or specific) when your NGS result place clustered reads right beside the primer pair.




@NestCommander - Kevin W. McCairn PhD
Since C-C say you need to @stevenemassey use qPCR to properly get the viral counts, let’s see…… Q61/Q70=PCR-. (And located uncomfortably close to PCR+ samples rendering them prone to contamination on NGS.) Q37=PCR- AND orphan sample negative whole stall before and negative exact site after. And primers aligned over by NGS. All are false positive samples. All does not prove virus is there with that metric. The virus is in the human+ and animal-poor Q64/Q68/Q69. What they wanted you to believe: Aerosols are blocked by walls and can not spread from toilets and wildlife stalls. Reality: Activity of samplers and vendors alike, especially their shoes and boots and the gloves of the samplers, caused the contamination to be spread out from the toilets. What they wanted you to believe: there are additional PCR+ samples. Reality: these are a different kind of PCR than what Jan 01 and Jan 12 used. It lacked lacked the universally present N primer pair in the specific PCR primers (the Jan 01 and Jan 12 used specific ORF1ab and N primers in the same reaction to generate 1 single Ct value) which indicate it being an non-specific (surveillance primers in PREDICT target only the ORF1ab/RdRp region due to its conservation, and have degeneracy.) test that cross react with all members of the Coronaviridae family. Artifacts ensues, if not “no reads at all”. Neither PCR+/NGS- nor PCR-/NGS+ can be trusted as genuinely positive, due to the extreme proneness to contamination in the NGS pipeline and the probability of cross-reactivity in some PCR tests. archive.md/2PM9Y archive.md/RirQ7 archive.md/CTP3i archive.md/NeybM archive.md/ETjzS archive.md/BWZJL https://pdfhost.io/v/~IGA2bONb_closest_to_the_toilets https://pdfhost.io/v/dUbkceTFh_anticorrelation_is_not_an_artifact And the reason why the samples in the market follow the rule which a positive sample archive.md/CTP3i archive.md/ETjzS archive.md/BWZJL must be contacted by samplers.🥼👖👢=positive. archive.md/NeybM archive.md/2PM9Y archive.md/RirQ7 And not frequently handled by vendors.🥩🥬🍄🛁📻☎️📦🧺=negative; Is the same reason why you only get animal viruses but not SARS-CoV-2 legitimate reads past 12/01/2020. https://assets.researchsquare.com/files/rs-885194/v1/78e0e6ce-4a76-48de-9f5c-76bab452bbe6.pdf?c=1665607885 Not found inside actual animal tissues because the animals are not infected, isolated SARS-CoV-2 virions are exceptionally sensitive toward destruction by RNAse 7 found on human skin, and they all got completely destroyed before the samples can even reach the testing lab if it contained material from a highly touched surface. Surfaces that see any hand contact at all in a hospital room just vaporized when the patient leaves, leaving only the floor behind which survive even terminal clean. Likewise, the skin surfaces of caretakers are free of RNA even when their stool become positive. Objects that always have either walking patient or HCWs touch like door handles keyboards or toilet seats have low prevalence even when the patient is inside the room, and objects which for a large fraction is touched only by gloved caregivers (bed rails) and objects that are strictly not allowed touching without a glove (ventilator buttons) when a patient is active, have the most SARS-CoV-2 RNA on them, where ventilator buttons which have 0% skin contact have much greater prevalence than bed rails which skin contact is absent in ICUs (where the positives came from) but present in normal wards. Similarly, heavily handled surfaces like old banknotes Are found to completely destroy SARS-CoV-2 RNA as little as 10 hours after deposition/material mixing to the same environment. Less handling and slightly longer life. Contrast clean surfaces like fresh PPE which the RNA remain stable for a month.




@NestCommander - Kevin W. McCairn PhD
https://assets.researchsquare.com/files/rs-885194/v1/78e0e6ce-4a76-48de-9f5c-76bab452bbe6.pdf?c=1665607885 in fact the nature of SARS-CoV-2 here being superfluous contamination that have an origin in human secretions and cell culture supernatants brought into the stalls by the samplers and on boots, shoes and suits is obvious here. Independent sampling indicate that only the animal viruses highly correlated with the animals were left in February. The SARS-CoV-2? Superfluous, in human metabolic products and secretions, and not in butchered tissues. They are cleaned off and degraded efficiently. The animal native viruses are inside the tissues and are highly persistent—class of contaminant type different and SARS-CoV-2 is in the class that is inconsistent with the behavior when pitched against cleaning than animal CoVs (which unlike SARS-CoV-2, the animal viruses including CoVs persisted in tissue fragments of animals generated from butchering and animals banging against cages for more than one month, whereas all the “SARS-CoV-2” after 12/01/2020 are either amplicon artifacts, PREDICT primer false positivities (no reads at all), or evidently freshly prepared cell cultures which even the fragile mitochondrial transcripts of the Homo Sapiens and intracellular viral transcripts of the SARS-CoV-2 have been preserved (they decay within a day after loss of cell viability https://www.ncbi.nlm.nih.gov/pmc/articles/PMC369693/) and where no non-human mammals at all were present. (Clearly a recent cell culture added into these “storehouse” samples). https://archive.md/13bdP archive.md/FskYn archive.md/gvHfw archive.md/4cCHG archive.md/csYBM archive.md/rj1pV archive.md/LJzSO archive.md/4cCHG boot on surface = NGS+/PCR+, suit on sample tube = PCR-/NGS+ as it indicate contamination occurred after PCR and before NGS especially when with alignment over the ORF1ab primer, and that the location then got a total negativity same stall before and same site afterward. Closer to the toilets, more likely of direct stall entry after market entry by the sampler, more samples become contaminated. Earlier the time of first sampling, the more virus in the contamination source at the entrance with less disturbance, and more virus is found in a sample that is taken from such a stall. And yes. One of the earliest unknown activity done by the WCDC including “taking environmental samples” and “cleaning” the market overnight in 31/12/2019. While the archive.md/iw1Pz animal samples have been disclosed in 01/2020 and all negative, the focus on early cases stalls in this run brought in the virus into the “live virus isolated” stalls that would be sampled in 01/01/2020.




@NestCommander - Kevin W. McCairn PhD
Trickery like using unauthorized sprayers to either put virus onto the stall (guess why all positive samples are below waist height? Yes. Sprayers are all aimed down by design and droppers can only be pointed downward when being used) immediately before sampling (resulting in NGS datasets containing transcriptomes of cells and virus that were far too fresh to be as old as a month and a half since the market is closed, if they have waited 11 days between putting the virus in and taking the samples that would have been able to weasel their way through) or that you put some additional Amplicons from another experiment-in-validation (keep in mind that the ORF1ab only primer pair as used in the “ORF1ab” PCR is distinct from the “ORF1ab/N” primer pair used before, as PCR kits of single Ct values are based on pre-mixed primer/probes that can not be separated for independent use, let alone “running out for just one primer”.) prior to running PCR in a sample (you get what you put in at NGS—amplicon only and other things that can react to the degenerate ‘ORF1ab’ primers but is not SARS-CoV-2 that are generated in your source experiment, but not legitimate SARS-CoV-2 reads) simply result in artifacts that clearly show evidence of sample manipulation within the resulting “data”. As the WCDC itself was also tasked to generate proof of whatever the most popular theory on zoonosis in that time, at first they attempted to just put human SARS-CoV-2 cultures into the wildlife stalls before a species is specified, yielding samples that correlated only With Homo Sapiens in a consistent manner or with significant mutual information. Then the primary suspect becomes “snakes” in that now debunked-by-ACE2 “codon usage” paper, https://onlinelibrary.wiley.com/doi/abs/10.1002/jmv.25682 And they were tasked to find a way to either remove or justify the human reads inevitably introduced with the virus. First they used their PREDICT primer pair (+SYBR green), which cross reacted but did not yield NGS SARS-CoV-2 reads. Then they tried putting one of their early WIP RdRp+multiple site amplification (one of the many different trials for suitable primer locations on the viral genome for distinguishing amplification prior to final probe design, already very close to their current N/E sites) experiments into the newly made “snake stall” samples, which supposedly exclude Human (some still snuck in alongside). The result are reads that are obvious artifacts that can not convince even untrained critics. Attempting to then justify the human reads if they can’t remove it, they went to the snake stall again and now took cultured virus straight from their incubators, mix it with “snake” meat samples impounded from earlier samplings (contained mixture of meats often sold as snake, but no mammals of any kind at all) thoroughly, put the result into the “storehouse” and immediately took swabs. Because of the immediacy of the action, the results are far too fresh by transcriptome to have been possibly deposited at or before 01/01/2020. (RdRp-based “ORF1ab primers” especially if there are other tests that are in development such as the “RdRp/N/E” tests are notorious for their cross-reactivity especially before the conditions are fully dialed in—the intermediate stage “ORF1ab only” primer https://assets.researchsquare.com/files/rs-885194/v1/78e0e6ce-4a76-48de-9f5c-76bab452bbe6.pdf?c=1665607885 also happens to be the primer used in the time when the animal CoVs are being identified in the market but not SARS-CoV-2 in their amplicons. The RdRp/N/E tests later generates 3 separate Ct values, indicating each test used 3 reactions likely validated separately, even on different sets of samples—https://www.sigmaaldrich.com/AU/en/technical-documents/protocol/genomics/qpcr/sybr-green-qpcr even primer dimers would react and that the command “take one amplified experiment each and put it into the environmental samples before PCR” would lead to legitimate read-free “ghost” reactivity, especially for experiments and primers that were still in early evaluation at that time. Same for contamination out of it.)





@NestCommander - Kevin W. McCairn PhD
(Note that samplers if they just came out of a lab doing specific PCR test development, viral amplicons and other complicated and not-all-in-1 experiments that involved the amplification of SARS-CoV-2 and other viruses, these can also drive contamination of the surfaces by the amplicons without deliberacy. Same if the workers have just attended to cell cultures in the lab—February Wuhan is actually among the time when PPE supplies especially the isolation suits and gloves/boot covers are in such a short supply that many disease control and hospital workers could only use one suit for an entire day—without being able to replace it even between tasks. You can expect that alongside sampling that got focused because of the snake theory, such compromising action to pull clearly artefactual intermediate-in-lab material out and into the environmental samples especially taken at the same day. Plus PREDICT primer cross-reactivity which this particular pair was used from 0127 to 0219.)
@NestCommander - Kevin W. McCairn PhD
@stevenemassey No. There is really no SARS-CoV-2 RBD in Wuhan.
@NestCommander - Kevin W. McCairn PhD
“Perfect synergy” that no live hosts wanted to keep. Proline: destroyed in all VOCs. Did not stop or prevent further animal infections with all the ones found infected before still commonly infected (no species were sold in Huanan). QTQTNS: became QTQTKS. Did not stop animal infections either, expands tropism in stead. The only things they are good in are in VERO/HAE and CaLu-3 cells.
@NestCommander - Kevin W. McCairn PhD
@stevenemassey It looks like raccoon dogs and civets are both entirely uninfected. Oops.
@RetractionWatch - Retraction Watch
“Criticism of statistical analysis on the origin of Corona.” https://www.faz.net/aktuell/wissen/medizin-ernaehrung/wo-der-corona-ursprung-wirklich-lag-fruehere-analysen-in-kritik-geraten-19476294.html


@NestCommander - Kevin W. McCairn PhD
@stevenemassey In fact, the entire “market centered” Chinese “early cases” data is tampered with and fraudulent.
@NestCommander - Kevin W. McCairn PhD
@stevenemassey HKU3 and ZC45 are not SARS1 or SARS2, nor were the “hubei civets” with Spike proteins nested well inside the Beijing strains of SARS1 a valid progenitor—it is a spillback infection. Nothing more.
@NestCommander - Kevin W. McCairn PhD
@stevenemassey And unfortunately, the “Hubei civets” are simply just spillbacks. And also, once again, ZC45r-CoVs=/=SC2r-CoVs.
@NestCommander - Kevin W. McCairn PhD
And even worse, the expected systematic social and populational biases from a survey of residences was too ignored within the China WHO “dataset”. An “meter precise” centering toward the market is exceptionally improbable from residences because they are too unevenly distributed around the market, biased populationally too toward the Wuhan CBD, meaning that even those infected near or are gathered from nearby the market should not have created an perfect centering of their KDE in a residence-free location of within 50m radius exactly above the market itself. To have this level of centering mean tampering with the “data” further, which leads to the majority of the “bullseye” cluster are not found on residential areas and that many of the “case residences” lands on water, which residences can not exist on without being washed away. Guess again why China never allowed any of the early case line list data or raw data of any kind to anyone? (And guess why they never dared to say where the first case they ever admitted or any cases at all lived at any later)?


@NestCommander - Kevin W. McCairn PhD
Not even rumors indicated any person at all in the wildlife industry in China being sick or getting infected, not even rumors indicated direct participation with the wildlife trade (purchasing, vending, dealing, transporting, farming, butchering, cooking or eating) by any of the known official or unofficial early cases. The only ever results from these wildlife trade participants indicate perfect condition of health and no evidence of infection at all among the customers or neighbors of any of them. Not even the market cases themselves—none of them reported direct participation of the wildlife trade. And unfortunately, The only thing governing the probability for positivity of the environmental samples is “closest to the toilets” and “closest to the main entrance of the market”. In fact, the samples in the market follows the rule which a positive sample archive.md/CTP3i archive.md/ETjzS archive.md/BWZJL must be contacted by samplers.🥼👖👢=positive. archive.md/NeybM archive.md/2PM9Y archive.md/RirQ7 And not frequently handled by vendors.🥩🥬🍄🛁📻☎️📦🧺=negative. archive.md/LJzSO archive.md/4cCHG boot on surface = NGS+/PCR+, suit on sample tube = PCR-/NGS+ as it indicate contamination occurred after PCR and before NGS with alignment over the ORF1ab primer. Closer to the toilets, more likely of direct stall entry after market entry by the sampler, more samples become contaminated. Earlier the time of first sampling, the more virus in the contamination source at the entrance with less disturbance, and more virus is found in a sample that is taken from such a stall. And yes. One of the earliest unknown activity done by the WCDC including “taking environmental samples” and “cleaning” the market overnight in 31/12/2019. While the archive.md/iw1Pz animal samples have been disclosed in 01/2020 and all negative, the focus on early cases stalls in this run brought in the virus into the “live virus isolated” stalls that would be sampled in 01/01/2020. https://wwwnc.cdc.gov/eid/article/10/6/03-0852_article On the contrast, 5 independent cases with close contact to the avenues of wildlife trade for SARS-CoV-1 have happened in 5 cities in 4 in Guangdong and 1 in Guangxi, over the same 2-months timeframe. Two of them were market workers on two independent markets which civets were sold, three of them were direct participants of the wildlife trade: two of them were civet butchers, and one a driver for wildlife dealers. All of these cases have yielded continued transmission from them. In the contrast, 0 of the early cases for SARS-CoV-2 worked in or have a history of direct participation with the wildlife industry. archive.md/yyX0Z archive.md/iw1Pz archive.md/4rVph archive.md/DChUL Exactly 0 raccoon dogs or any of the so-called “susceptible species” were found infected anywhere in the world, not even by a relative of SARS-CoV-2.




@NestCommander - Kevin W. McCairn PhD
Again, asking: why they have to ban the Wuhan P4 lab from mentioning in 31/12/2019, before any theories can even be made? (Also notice that the first market case neither worked with the wildlife trade, nor did she even play mahjong like the later cases did when the linked cases were first gathered by a citywide command for them over 30-31/12/2019. Once again indicating that the market outbreak was likely first brought in from the outside, then superspread at the toilets and mahjong rooms later.)




@NestCommander - Kevin W. McCairn PhD
Why Daszak’s name ended up in the GenBank file of a—Laotian CoV, which they claim to have never sampled especially alongside Shi as well? “ID’ing 50+ novel strains”. There is Inconsistency piled upon inconsistency in Chinese publications as well as “data”. And he just held onto his secrets for 9.5 hours and also, notably, failed to produce a counterargument, after 9 and half hours. The ODNI broke the law and their “report” is full of elementary mistakes. More hidden experiments.




@NestCommander - Kevin W. McCairn PhD
Also, the only bat cave that was banned from public access—were Mojiang and Shitou. It happens only at and serves only to obscure the actual inventory under their control in the sampling and testing sites of the WIV. And they Did not “Stop all bat sampling”. There is clearly evidence that they are perfectly capable of hiding their work, none of the claimed official audits to the lab was ever published or even disseminated among the authorities in China, And the reason why they used GISAID here, is that https://archive.md/0aHWr https://archive.md/Myt4u there is no proper versioning or custody of “data” on GISAID. https://archive.md/52DyQ https://archive.md/B0xlW @DiLiMengYAN1 And no trails that can be FOIA’ed and bust their “data” like on NCBI. And no possibility that inconvenient lab-incriminating data can be leaked or FOIA’ed like csabai et al. Still no official response from the CCP.
@NestCommander - Kevin W. McCairn PhD
“They would close the lab and raze it to the ground”: That is admission of guilt, not cover-up. What they actually did: forged the environmental samples by spraying the wildlife stalls with the virus in Jan 02. Erased the superspreading site of the toilets in Feb 13. Refused to swab anywhere in Wuhan outside the market or its immediate vicinities, not even other corners of the market. Shoved all the cases with original residence in Wuchang into the market. No government-funded lab have been shut down after a leak, even when outbreak and outrage ensued. The interest in keeping the labs active and operational is that of national security, keeping the biodefense industry stable and up-to-date. They are never shut down, only repaired usually with as little disruption or outside visibility as possible. (WIV had an 2 year batCoV research publication hiatus, despite attempt to keep mitigation efforts secret). No civilian interests can interrupt the operation of these labs. https://www.thefreelibrary.com/Farmers+demand+a+Pirbright+shutdown%3b+%27A+private+company+would+have...-a0168453941 Not shutting down after leak is also one of the decisions that well, even democracies, does. and Yes. Pirbright is back at FMDV again after the leak. “Does this look like what a lab would do after leak causing hundreds of millions of pounds of damage”? https://en.wikipedia.org/wiki/2007_United_Kingdom_foot-and-mouth_outbreak Fact: Pirbright was not shut down after FMDV leak infecting 4 farms nearby. They repaired their drain pipes and continued operation, not even interfering academic publication patterns. Ironically, cow farms were shut down and beef trade was closed during the outbreak. This resulted in an epidemic lasting 5 months in cows that lead to at least two major cullings and severe disruption to the livestock trade from the U.K. That is, the reaction look like what they claim a zoonosis would look like, not what they declare what the WIV would do when such a shut-down would certainly directly admit guilt and spell doom to both the institute and its operators. https://www.theaustralian.com.au/science/beijing-lab-mishap-infected-scientist-with-covid19/news-story/9b0cb0ed84df21d25da11b698be3611a Fact 2: there is no shut-down reported at all in the IVDC either after the 2004 leak of SARS or the 2020 leak of Covid. Not even a burp of interruption. Fact 3: the WIV went on hiatus to the bat CoV isolation tests over 2020-2023. When the sverdlovsk anthrax leak happened, they blamed the animal farms and markets nearby and did not officially shut down the facility. The construction of another anthrax facility nearby was considered potential indication of a shut-down, which is on par with the WIV hiatus. After a timescale similar to the WIV hiatus, the new facility was opened for inspection which no anthrax was found, meaning that they fixed sverdlovsk and went on, just like the WIV (chen WEI……). In facts, there have not been a single record of an lab leak or LAI in a research facility that resulted in the (especially permanent, as what they claimed would happen) shut down of the facility (despite hundreds of known incidents in record), even when significant epidemic have occurred from the event. (Ebola21, FMDV07, H1N177, Anthrax82 which no official shutdown was known).

@NestCommander - Kevin W. McCairn PhD
there is a long history of the WIV lying to the point of base rate neglect when being asked anything about potential LAI. The “dinner of staff” too, where they neglected the base rate which is Wuhan medical institutions are already in panic and the general public is already taking precaution, as h2h is announced in 15-16/01/2020 to the point that even the invited international collaborator have hinted Shi to wash hands, that she unexpectedly did not given her expertise and knowledge on the public info about SARS-CoV-2 in general Wuhan public in this time. She pretended to not know the need to take precautions when she was expected to do so, just like when she sabotaged the test to make 67 general 2021 Wuhan public serological samples test all negative when there should be positives given the seroprevalence in Wuhan at that time. researchgate.net/publication/35… It is just as impossible To have 67 community members to test all negative in Wuhan in 01/2021 as to have 593 people to test all negative with any sensitive test available in April-June 2023. gab.com/Flavinkins/pos… And this same behavior of issuing a test that will not turn positive on a human also happened to the mojiang miners. Where their own early serological test results were contradicted. archive.md/Pc6gp archive.md/zUD1F And ben HU lied about working with live virus which are so easy to debunk just by a simple google search. His own grant notice required live virus work in 2019.
@NestCommander - Kevin W. McCairn PhD
Importantly, none of the species in that “stall” were truly susceptible and 0 individuals of any of the species have been observed with an infection with a relative of SARS-CoV-2 in the wild. “Susceptible species” is nothing but a myth and this reflect well by the fact that None of the “susceptible species” were actually in positive correlation at all with the SARS-CoV-2 reads once you enter that “stall”—it is confounded by the toilets and all it had in it is sampler contamination, just like the outside of stall W4-26-28. archive.md/gvHfw archive.md/csYBM archive.md/vlAgp archive.md/DChUL archive.md/4rVph archive.md/yyX0Z Despite surveillance in Europe and Japan, there were zero evidence of natural infections in a raccoon dog anywhere in the world. archive.md/iw1Pz All upstream suppliers are traced and were negative. In fact, none of the “susceptible species” have evidence of even a single individual being infected by SARS-CoV-2 or its relatives anywhere in the world. archive.md/VNr75 archive.md/rj1pV All 3 lineage A samples have direct link to the WCDC. The stall of A20 have owners that Wore slippers and handle fish with bare hands. They don’t wear gloves and shoe covers can’t be worn over slippers. This sample is contamination caused by the WCDC itself and there is no case from the market that is lineage A. This same infected sampler, that the WCDC would even admit, would then go on sampling the wildlife stalls in Jan 12 and rub his contaminants all over the surfaces on the closest stall to the toilets and sample tubes. This then drive confirmation bias on snakes which lead to more sampling and more contamination, all contained nothing but artifacts and never on the original site of the contaminated sample tube again, eventually leading to samples with only human DNA and no other mammals at all inside. Zero attempts have been made to gather evidence at the WIV. https://pdfhost.io/v/kZ1ilPCFa_The_real_problem_is_that_there_was_literally_zero_attempts_at_gathering_any_evidence_at_the_WIV Or anywhere else. https://pdfhost.io/v/dUbkceTFh_anticorrelation_is_not_an_artifact And the anticorrelation with raccoon dogs aren’t “an artifact”. The “all samples” correlations are artifacts of pathological spatial distribution from confounding factors that extracted the species found in the least number of overall stalls that happened to include the stall closest to the toilets and entrance into the market during the sampling run. https://pdfhost.io/v/~IGA2bONb_closest_to_the_toilets This mean that all correlations other than humans failed when the analysis is to be done in a way which the pathological spatial distribution is mitigated in any way. archive.md/MtkL3 All species other than humans at most landed on entirely different sections of the ground and set of items than the SARS-CoV-2 reads, and only humans are found on the same sections of ground and set of items in the form of sampler-linked contaminants. This is evident when the correlation is performed with only samples where SARS-CoV-2 is found, which effectively queries “which species shed the SARS-CoV-2 sequences in samples where it was found” as opposed to “which set of species most uniquely represents the spatial features of the single stall closest to the toilets”. Hedgehogs have proven non-susceptible ACE2. Oh. On the entire “early cases map”: @CharlesRixey The entirety of that “map” was created using concocted and fake “data” spoon-fed by China that wasn’t available to the public even this date. archive.md/5sdkR archive.md/1pcCU archive.md/N0hib archive.is/Kyr1z archive.md/VXtu9
@NestCommander - Kevin W. McCairn PhD
@AntGDuarte And here is a hint: despite being found all over the stalls and were also the objects most handled by the human cases, even in stalls with human cases and where there were no wildlife DNA in the positive samples from the stall, no boxes or baskets in the market have tested positive. The same for cashiers, keyboards, monitors, water cups or any objects that are frequently handled by direct touch by a vendor on the surface where the swab will be taken from. They can never test positive because of the insanely high content of RNAse 7 and other defensive nucleases on human skin destroys SARS-CoV-2 virion RNA within the period which the samples are stored inside liquid medium before being tested (which is particularly effective when the RNA is found in loose virions or human metabolic secrations and not shielded inside solid animal tissues), and vaporizes archive.md/RirQ7 archive.md/CTP3i archive.md/NeybM archive.md/2PM9Y any virion-free RNA the instant it enters medium contaminated by it. The WCDC and the Hubei CDC stores all of the human samples and backups of research cultures of pathogenic microbes in Wuhan, as this is their legally delegated duty (the “各级疾控部门” are termed “保藏机构” for “病原性微生物” under Chinese law governing the use of cultures and samples of human and animal pathogenic microbial samples, and samples that were suspected to have the possibility of containing such microorganisms. These are also the only locations which first round samples arriving in Wuhan are allowed to go for pre-screening prior to entry into the other labs in Wuhan, “检测机构”. ) and that labs in China are not allowed to store such cultures except several select state key laboratories. Since 2014, the only EID surveillance target in Wuhan is the HSM which all other sites are kept blind so that they can blame Huanan in case the research labs suffer an accident. Almost the soon as experimentation begun in the WCDC at the first detection of an infection from that program (Chen/WIV), the prior culture samples that was identified to match (via preliminary testing, including RdRp and antigens which are targeted by the Military test kits used in Wuhan) ended up causing an employee infection. Creating all 3 lineage A cases afterward. The employee infection would end up being detected because the CDC have to use a kit that work on real patients unlike the WIV, and got whistleblown into the WHO report under the pretext of an again never-specified “family cluster transmission”. So bad that you can’t actually compare Serological tests that were conducted between distinct times and groups of people for the test itself because doing so violated the statistical homogeny criterion for test efficacy evaluation—the same as comparing 🍎 with 🍊.
@NestCommander - Kevin W. McCairn PhD
Reality 1: many different supply chain exist from Yunnan to Guangdong and Hubei is just infected in SARS1 by human cases. Reality 2: The actual count for animals farmed in China vs sold in Wuhan likewise indicate that Wuhan sell only a negligible fraction of all animal sales in China Especially when compared to Guangdong.
@NestCommander - Kevin W. McCairn PhD
https://www.nytimes.com/2021/02/12/world/asia/china-world-health-organization-coronavirus.html They systematically moved more than 3000 cases from the lab to the market and gave “cases data” that they wanted to push for market as first outbreak site to distance from the labs. https://archive.md/rYvu3 https://archive.md/UFrSv https://archive.md/nevZy https://www.researchgate.net/publication/370635299_Greater_than_the_Sum_of_its_Parts_-_Aggregated_Wuhan_COVID-19_case_data_points_to_the_wrong_side_of_the_Yangtze_River_-_Rixey_-_20230509 Such an result of having unlinked cases closer to the market than linked cases is not expected even under the null hypothesis of market origin, which we should see unlinked cases secondary to and cluster around the linked cases, and not the market itself. https://www.researchgate.net/publication/370635299_Greater_than_the_Sum_of_its_Parts_-_Aggregated_Wuhan_COVID-19_case_data_points_to_the_wrong_side_of_the_Yangtze_River_-_Rixey_-_20230509 Not only there were an complete absence of verifiability in Chinese cases, there is direct non-circumstantial evidence that they moved up to 3000 cases from Wuchang to Huanan. In fact, it is totally not normal to have unlinked cases closer to the market than linked cases—the only way this can happen is with ascertainment bias. Only near the market gets ascertained if not directly linked to it. Base rate neglect. They did the exact same thing when claiming that all 67 “pre-Huanan checkable cases” were “serologically negative”. Again, the social media associated here say “before Jan 18, 2020”. Included all Dec cases. https://www.mdpi.com/2220-9964/9/6/402 Before they begun enforcing their claim of “100/174 centered around the market” and starting to tamper with data to make the claim, https://ghrp.biomedcentral.com/articles/10.1186/s41256-021-00200-8 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7149375/ 135/92 and 115/82 cases already got into in early peer-reviewed papers that went missing in the WHO report. Past media reports archive.md/Ea0Kw archive.md/1x658 also contradict WHO in key early cases’ residences, including the earliest case they admit in the WHO report. archive.md/5sdkR archive.md/1pcCU archive.md/N0hib archive.md/VXtu9 archive.is/Kyr1z https://archive.org/details/mace-e-pai-covid-19-analysis-redacted/page/8/mode/1up And you know that they hate this information when it was censored. The MACE-EPAI document here is not searchable on google. Up to one third of all cases were either removed completely or moved toward the market in the “dataset”. archive.md/zUD1F archive.md/Pc6gp https://archive.is/p3K3Z Including the very first case they ever admitted officially. And outright removed 4 times more cases than official. Unlinked cases supposedly secondary to linked cases should cluster around them, not the market itself. archive.md/GvRcD archive.md/ZgVzp Wuhan authorities after that archive.md/OIGPz 2014 incident now targeted only the Huanan market when looking for EID outbreaks—and nowhere else. archive.md/1x658 They tampered with the early cases data archive.md/Ea0Kw To make it look like it “started at the market” when in reality the first case they ever admitted lived right next to the WIV BSL-4. archive.md/5sdkR severe discrepancy happening December 2019 and January 2020 indicate tampering with case counts. archive.md/1pcCU This is indicative of catastrophic ascertainment bias was going on. None of China’s “early cases” dataset is credible. https://archive.md/ET1GA https://archive.md/Ea0Kw https://archive.md/1x658 The tampering of early case residence data is systematic and extensive. It is the reason why they refused to provide this data in any detail at all.
@NestCommander - Kevin W. McCairn PhD
Not only did The first every case they admitted live in Shidong right next to the BSL-4, and were moved toward the market in the WHO report in contradiction to all known media coverage, https://gab.com/Flavinkins/posts/109256201942085712 the entirety of Wuchang district was wiped clean for every single WHO case that have onset before 27/12/2019–with up to 3000 cases moved to the market this way over the entire Wuhan outbreak. https://archive.md/1x658 and for central Wuchang near the labs and the densest inhabited regions inside the district, all cases were moved away in the WHO map. Unfortunately Rasmussen's work on the origins question rests heavily on what David Relman described as "hopelessly impoverished" early case data. https://www.washingtonpost.com/national-security/2023/02/27/little-known-scientific-team-behind-new-assessment-covid-19-origins/ https://www.washingtonpost.com/opinions/2022/11/17/covid-early-cases-wuhan-china-mystery/ https://archive.md/ke1lp https://archive.md/RaYPC David Fisman: I think the most interesting thing this fellow says is that there are clearly tens of thousands of cases...That implies a much earlier introduction than would have occurred with a seafood market outbreak..." Also, Chen is not the only person infected in Shidong/Jiangxia and central Wuchang. Most were censored and only one of the two ambulances arriving in 31/12/2019 have been registered as a dot—likely because the origin wasn’t inside the Shidong prefecture/BSL-4 surroundings, and likely only because of being a close contact relative of Chen (contacting an known case). https://gab.com/Flavinkins/posts/109256201942085712 All dots they moved this way (up to 1/3 of all cases) was sent to Jianghan, https://archive.md/p3K3Z https://www.researchgate.net/publication/370635299_Greater_than_the_Sum_of_its_Parts_-_Aggregated_Wuhan_COVID-19_case_data_points_to_the_wrong_side_of_the_Yangtze_River_-_Rixey_-_20230509 especially to the immediate surroundings of the market, to scapegoat it and end up causing the “unlinked cases” cluster to be closer to the market than the “linked cases” cluster, despite supposedly the linked cases should be the only source of initial human to human transmission seeding and therefore the unlinked cases should cluster near the linked cases and not the market itself. This kind of improbable-under-null-hypothesis behavior is all over Chinese “data”. archive.md/VNr75 archive.md/rj1pV They attempted to spray their culture into the wildlife stalls, which ended up Making Homo Sapiens the only species that is found in every sample with a viral read in the market (note the absence of lineage reads in the wildlife stalls), and archive.md/LJzSO archive.md/4cCHG archive.md/13bdP all of the subsequent efforts at creating positive samples where the CCP specified them to do (“Blame snakes!” Is the official voice in 02/2020) just brought in artifacts first, and then when all of the mammals have degraded away, pure cultures of SARS-CoV-2 intracellular transcriptomes in human cellular transcriptomes. In addition to the heavy censorship of case ascertainment effectively mean you have to either live near the market or have a direct or indirect link to be diagnosed at all, moving all Wuchang case residence dots and sending them to Jianghan archive.md/1x658 archive.md/Ea0Kw also caused the “unlinked” dots to cluster closer the the market than the “linked” dots—something that can not happen without data manipulation on a massive scale. https://archive.md/ET1GA Unlinked cases are supposed to be seeded only by the linked cases if they didn’t visit Huanan under the market origin assumption. They are supposed to cluster near the linked cases and NOT the market itself. The CCP failed in this elementary logical analysis and resulted in a “dataset” that is too perfect to be possibly real. https://gab.com/Flavinkins/posts/108830214433800007
@NestCommander - Kevin W. McCairn PhD
@Florin_Uncovers - Florin
So the fraudulent coder in Pekar et al. '22 @niemasd ran Samson et al. '24 data and CONFIRMED their Aug-early Oct '19 tMRCA result!😅 Then he did what @michaelworobey & @jepekar did: discarded inconvenient data not realizing you also need animal sequences to model a bottleneck!🤡

@NestCommander - Kevin W. McCairn PhD




@NestCommander - Kevin W. McCairn PhD
Once again, 1: RaTG13 is not viable. https://zenodo.org/record/5702700#.ZJ2KiyV6slT https://zenodo.org/record/5778318#.ZJ5hyCV6slT 2: the real issue is that 1. WIV lies about everything serological. None of their “tests” were positive when politics require it to be negative. And 2. The missing sequences of Latinne et al is where you find what the WIV was working on. 7 SARSr and 54 total CoVs were missing entirely.




@NestCommander - Kevin W. McCairn PhD
https://archive.md/OIGPz The “Shunde problem” or “why it managed to infect Wuhan and only Wuhan”—is a problem which all market zoonosis or wildlife farm theories require extremely improbable and hard explanation to answer. Unfortunately the actual sales of wild animals in 2019 contained metadata-supported images or videos only in Guangdong and Guangxi, and not Wuhan. All observations of virologists working at the market without a published sample taken at that date should automatically be considered extremely suspicious. The most likely reason is that They were dropping in samples in stead of taking them, leading to the observation that only human have a consistent positive correlation or any significant mutual information with SARS-CoV-2 there. The reason why China intentionally hid nearly half of their flow cells is because they could use the reads inside to tamper with the “wildlife stall data” to meet the demand of the zoonati when given in 11/03/2023. They used it to scramble the host counts in all their “negative samples” when the correlational edge with Homo Sapiens were found to persist despite they removing the 300nt+ non-viral contigs and leading to an inverse correlation between the residual mitochondrial singletons * SARS-CoV-2 and the leftover contigs of other mammals as they were shredded by the common 43nt nuclear reads inside all mammalian genomes. Even before that, to prevent the obvious and embarrassing conclusion of “the SARS-CoV-2 is most likely smeared out of the toilets by the samplers” when both Jan 01 and Jan 12 have the stall with most positive samples turned out to be the one that is closest to the toilets and where the samplers entered and existed and a national plan was made to sample the toilets and public activity rooms in response, Wuhan ordered the bleaching and destruction of the toilet area before a sample can be taken from it. In fact, the civilian side of the national disease control apparatus was not even allowed to see the Q* samples in person or sequence them independently—They were not even allowed to verify any of the “qPCR results” and not even an Ct value would be “reported from the lab” which sent in the “sequencing results for Q* samples” directly. Eventually Xi ordered all Covid-relevant Departments to follow the same operational instructions over the end of February to the beginning of March 2020, the point of which an agreement was finally struck that they would work together to fabricate a “dataset” for animal origins, first as the primary (rewards were handed out to “find the animal origins of SARS-CoV-2” as late as 05/2020, alongside numerous NCBI data replacements and changes that happened over 02-04/2020 on all of the bat and pangolin datasets for “animal origins” leaving behind corresponding artifacts) and then as the fallback plan after 05-06/2020.




@NestCommander - Kevin W. McCairn PhD
Deliberate “Spicing up” of samples by Wuhan. Note how not even informal sources out of China have published what samples were taken in 02/01/2020, or even acknowledged the performing of sampling work in 02/01/2020 (which the existence of intensive virology-related work at the market, particularly focusing “around W7” e.g. from w6 to w8, was known only by eyewitness account by outsiders but not any official or informal acknowledgment by the operators). (Unlike 31/12/2019 which the performing of sampling work was acknowledged by the WHO report and Jiangwei, which archive.md/yyX0Z archive.md/iw1Pz archive.md/4rVph archive.md/DChUL the explicit collection of animal samples at that point ended up as negative test results that were disclosed in private channels in January 2020.) And all those trampling by contaminated boots and rubbing by contaminated suits(potentially even contaminated gloves, which unlike bare vendor hands start sterile and RNAse-free, and touches mainly surfaces just above step height as aseptic techniques becomes progressively more difficult to uphold when virology operations are performed while bowing down) are going to cause extra contamination, out of the toilets and in from then outside, particularly on all places especially those that are heavily trampled, inside this area, that were not then cleaned prior to sampling later. Reason why the only consistent positive correlation or significant mutual information between SARS-CoV-2 and species is Homo Sapiens, Despite read filtering and data obfuscation especially at that time. (No independent validation, missing method details, sometimes not even Ct values were allowed to Gao et al, only what Wuhan claimed they did and produced exclusively in-silico)




@NestCommander - Kevin W. McCairn PhD
https://archive.md/VXtu9 The actual R0 and serial interval is much, much lower and longer, contaminated by change of ascertainment criterion. Different strains spread differently. Coronaviruses superspread instantaneously and not spread continuously as HIV which FAVITES bases on. https://gab.com/Flavinkins/posts/108860074766577121 There is an inverse polytomy size to time in SARS-CoV-2. VOCs are bigger than B.1. B.1 is bigger than B. B bigger than A is expected. Unfortunately, B is in fact more transmissible and mutate faster than A…… (reason why A went extinct, and also skewed the tMRCA analysis) There is nonlinearity and an infected brain to boost. The entire assumption for pekar et al is wrong.

@NestCommander - Kevin W. McCairn PhD
Not even rumors indicated any person at all in the wildlife industry in China being sick or getting infected, not even rumors indicated direct participation with the wildlife trade (purchasing, vending, dealing, transporting, farming, butchering, cooking or eating) by any of the known official or unofficial early cases. The only ever results from these wildlife trade participants indicate perfect condition of health and no evidence of infection at all among the customers or neighbors of any of them. Not even the market cases themselves—none of them reported direct participation of the wildlife trade. And unfortunately, The only thing governing the probability for positivity of the environmental samples is “closest to the toilets” and “closest to the main entrance of the market”. In fact, the samples in the market follows the rule which a positive sample archive.md/CTP3i archive.md/ETjzS archive.md/BWZJL must be contacted by samplers.🥼👖👢=positive. archive.md/NeybM archive.md/2PM9Y archive.md/RirQ7 And not frequently handled by vendors.🥩🥬🍄🛁📻☎️📦🧺=negative. archive.md/LJzSO archive.md/4cCHG boot on surface = NGS+/PCR+, suit on sample tube = PCR-/NGS+ as it indicate contamination occurred after PCR and before NGS with alignment over the ORF1ab primer. Closer to the toilets, more likely of direct stall entry after market entry by the sampler, more samples become contaminated. Earlier the time of first sampling, the more virus in the contamination source at the entrance with less disturbance, and more virus is found in a sample that is taken from such a stall. And yes. One of the earliest unknown activity done by the WCDC including “taking environmental samples” and “cleaning” the market overnight in 31/12/2019. While the archive.md/iw1Pz animal samples have been disclosed in 01/2020 and all negative, the focus on early cases stalls in this run brought in the virus into the “live virus isolated” stalls that would be sampled in 01/01/2020. https://wwwnc.cdc.gov/eid/article/10/6/03-0852_article On the contrast, 5 independent cases with close contact to the avenues of wildlife trade for SARS-CoV-1 have happened in 4 cities in Guangdong and 1 town in Guangxi (+1 city which contact is unknown), over the same 2-months timeframe. Two of them were market workers on two independent markets which civets were sold, three of them were direct participants of the wildlife trade: two of them were civet butchers, and one a driver for wildlife dealers. All of these cases have yielded continued transmission from them. In the contrast, 0 of the early cases for SARS-CoV-2 worked in or have a history of direct participation with the wildlife industry. archive.md/yyX0Z archive.md/iw1Pz archive.md/4rVph archive.md/DChUL Exactly 0 raccoon dogs or any of the so-called “susceptible species” were found infected anywhere in the world, not even by a relative of SARS-CoV-2. archive.md/GKdtc https://archive.md/e3615 https://archive.md/vWjZl https://archive.md/nyR0q China did not put any real ban or even influence on the wildlife trade at all especially Guangdong, before the beginning of 02/2020. The first market case is in 11/12/2019. In the ~2 month time window, all 5 of the “directly wildlife linked” index SARS1 patients have already been infected. And more than half of the 11 known index SARS1 patients, over 5 of the 9 index locations. Official denial of wildlife trade did not at all influenced the real trade that was happening, which in Guangdong also proceeded all the way to the Chinese new year of 2020, which is well into February. There is no evidence at all that there is a sufficiently timely ban of wildlife trade in China to stop all and every secondary spillovers especially Guangdong.

@NestCommander - Kevin W. McCairn PhD




@NestCommander - Kevin W. McCairn PhD
archive.md/DChUL archive.md/yyX0Z archive.md/4rVph archive.md/iw1Pz https://pubmed.ncbi.nlm.nih.gov/35298912/ https://pubmed.ncbi.nlm.nih.gov/35298912/ In fact, the raccoon dogs are locally wild-caught within Wuhan, that human Herpesvirus is identified indicating human contamination have occurred alongside the clearly unique human mitochondrial reads identified, and that there are zero mutual information in term of read abundances between SARS-CoV-2 and the animals. Only the human mitochondrial reads. Worse—all of the animal species correlated perfectly with their expected viruses, and the only species which SARS-CoV-2 is the perfectly correlated expected virus is “Toilets and Homo Sapiens”. In fact, The only thing governing the probability for positivity of the environmental samples, the so-called “spatial correlation”, is “closest to the toilets” and “closest to the main entrance of the market”. Spoilers: the actual stalls that sold animals from Yunnan are entirely uninfected. It is entirely expected with zero evidence of even a single SARS-CoV-2 case linked to any of the intermediate distribution sites and secondary destinations even in Hubei or wuhan of any of the animals that were supplied to the Huanan market, especially given that the each stall have at least 3 distinct live animal suppliers for “susceptible animals” and there are 17 stalls in Wuhan, and the total number of animals sold per week is only ~58 in total. 4 animals at most per shelf life per supplier is not going to eat up the single harvest output of any farm. It will spill into other cities. None observed.





@NestCommander - Kevin W. McCairn PhD
Issue: the drains don’t actually have SARS-CoV-2 reads inside. Only persistent, cross-reactive animal CoVs and potential trample marks. Putting bleach onto the toilets also doesn’t help at all.


@NestCommander - Kevin W. McCairn PhD
Unlike animals including livestock, humans are neither sold nor butchered at the market. Their CoVs degraded catastrophically after 01/01/2020 and completely after 12/01/2020 leaving only artifacts behind. archive.md/13bdP archive.md/FskYn archive.md/gvHfw archive.md/4cCHG Animals that are sold and butchered at the market have their CoVs remain stable and are the only CoVs left detectable in February 2020. (note there is a continuous deposition of ratCoVs due to the rats that ran through the market nearly daily after closure (they begun to show only after 12/01/2020 when rats begun to severely infest the market). There is no possible deposition of SARS-CoV-2 or other animal CoVs by nonsampler sources after the closure of the market.(The animal CoVs that are not RatCoVs were found with consistent counts over Jan01, Jan12 and all later dates. SARS-CoV-2 rapidly decline from Jan 01 to Jan 12, then are completely gone leaving no reads that isn’t an obvious anatrifact later) This https://assets.researchsquare.com/files/rs-885194/v1/78e0e6ce-4a76-48de-9f5c-76bab452bbe6.pdf?c=1665607885 further exemplified the fact that the SARS-CoV-2 found in the market here is superfluous contamination that is distinct from the animal viruses or CoVs. One fact among many that disagree with animal origin. https://t.co/VV9Gzg7JKi? And spoilers: none of the “susceptible species in W6-29-33” (wild species that is found there and have not been rejected as unlikely susceptible experimentally) garnered a positive theil-sen estimator result in any of the slices examined. This is in addition to the fact that meaningful correlation especially ones with significant mutual information was found to animals only with animal-specific viruses, and SARS-CoV-2 to only Homo Sapiens. Why ignore the toilets again and again? W4-26-28, especially W4-28 where only 1 out of the 2/2 human cases-free positive sample have anh wildlife DNA, have the exact same cause for maximal positivity in Jan 01 as W6-29-33 in Jan 12: closest to the toilets. @jbloom_lab If you think 20 ILI samples per month can isolate the one covid case in a sea of 8000+ flu cases every two weeks. Or that pre-screened pack tube blood verified at banking to be IgM free can detect the ~100 SARS-CoV-2 IgM+ cases expected in November 2019.
@NestCommander - Kevin W. McCairn PhD
Were any corner of the WIV or even the Hankou station itself ever sampled? And well—Yunnan and Guangxi animal stalls were—Completely uninfected. https://archive.md/p3K3Z And Up to one third of all cases were moved from the lab to the market. Dazhong stopped its wildlife sales in 2014. Negative. The Yunnan and Guangxi animals are sold in W9-34-36 and W8-36-38. Negative. In deed, this is a spurious result—just like how entering the stall which you find the SARS-CoV-2 and the correlation crashed with the porcupines but kept that with the humans, theil-sen correlation give no mutual information at all to those pocrupines except a negative one with samples where SARS-CoV-2 is found. And of course, if you think that someone sick in November 2019 would not be able to meet in 15/12/2019 when the max length for sickness is merely 15 days for younger people…… Any susceptible species with significant mutual information at all and Homo Sapiens have the max mutual information. Animal CoVs are consistent in all dates not just Jan 01 and Jan 12 including after Jan 12. SARS-CoV-2 reduces rapidly in concentration from Jan 01 to Jan 12, and disappeared after Jan 12. Not inside sold animal tissues=rapidly degraded by RNAse 7. They scrambled all mutual information in 26/03/2023. Transfer contamination from the sampler labs and the toilets account for all market samples. Not vendors or animals.
@NestCommander - Kevin W. McCairn PhD
And of course, there isn’t really that an “connection” when you realized that all the “Hubei SARS” strains are in reality just HKU3 and ZC45 none with even the right RdRp or RBD, and thus the raccoon dogs, the very raccoon dogs that were being shipped to the HSM for sale, are just as expected, entirely negative at testing.




@NestCommander - Kevin W. McCairn PhD
Initially, the toilets and the sampler pants and boots smeared the contamination out into the stalls, leading to the first set of samples which the stall with most positives samples out of all samples being always the stall that is closest to the toilets. At this time, they have also attempted to spray the stall with animals and virus as in Jan 02, when an army of hazmat suited workers performed virology work which no official or unofficial accounts for performing the work as sampling in that date was known, were identified by eyewitness records. Because the animals are all museum specimens that were far too dry to properly resuspend, the first attempt at faking “animal origin” ended up with A total absence of any consistent positive correlation or significant mutual information at all Between SARS-CoV-2 and all species other than Homo Sapiens. You can easily distinguish between common tertiary cause (confounded) from true causation by looking with increasingly finer grain of resolution, especially where the data points aren’t 0. Unlike spurious correlations from Confounding factors which ends at the resolution where the factor acts on, True causation stay correlated in every resolution and in any set of data points especially where the data values aren’t 0. In fact, confounding factors often crash in correlation quite early before that. The PREDICT ORF1ab only (RdRp) primer was used in stead of the initial “ORF1ab/N” primer set, between 27/01/2020 and 15/02/2020. https://assets.researchsquare.com/files/rs-885194/v1/78e0e6ce-4a76-48de-9f5c-76bab452bbe6.pdf?c=1665607885 The resampling of the wildlife stalls in the beginning of February 2020, within the same period, resulted in only animal-specific CoVs but no SARS-CoV-2 when amplicons generated with this primer pair were sequenced. archive.md/VNr75 archive.md/rj1pV archive.md/LJzSO archive.md/4cCHG Then, the leading hypothesis becomes snakes due to the “codon usage” paper, https://onlinelibrary.wiley.com/doi/abs/10.1002/jmv.25682 and as usual, in the continued attempt of fabricating “evidence” for whatever leading hypothesis at this time, they tried multiple ways to eliminate the human correlation edge of their initial products and dramatically oversampled their “snake stall”. They first started using Oligonucleotides and amplification products from their developing “RdRp/N/E” assay, resulting in artifact-only NGS alignments and no reads at all as these products including primer dimers generated off the test being developed and other, failed PREDICT amplification experiments, contaminates the boots, Suits and gloves of the samplers in one run and all the sample tubes used in another, with PPE in Wuhan at that point so scarce that workers often have to use the same suit for the entire day between lab work and sampling. When the E and N amplicons are present in the amplification product used, they show as single-amplicon artifacts. They also attempted verify their Q37, which the snake stall tested negative in Jan 01, but all results are failures. Facing the issue with either cross react or primer dimer and get nothing, or viral amplicon and get only amplicons with their “adulterate with amplification products” attempt (the only drain with a real SARS-CoV-2 read is a municipal sewage well on the opposite corner than the wildlife stalls!); archive.md/13bdP They decided to take samples of fresher meat from the market (animal sampling have begun in this time) that included snakes but failed to include any mammals, blend it with cell cultures and spray it onto their final sampling site “storehouse”, hoping that this would equalize out any edge humans have in correlation. The cultures https://www.ncbi.nlm.nih.gov/pmc/articles/PMC369693/ ended up far too fresh for the purported deposition date of pre-Jan01, and when the snakes are debunked, confirmed to be pure artifacts.
@NestCommander - Kevin W. McCairn PhD
And also, regarding that so-called “nature’s GOF laboratory” claim—to this date, zero Sarbecoviruses with an FCS have ever been identified. web.archive.org/web/2022101805… web.archive.org/web/2022090222… In natural settings, an animal will seroconvert before the FCS can emerge, which is extremely unstable especially in D614 inside seroconverted hosts. In fact, this prevented the FCS from emerging even inside the 2002-2003 SARS-CoV-1 in the exact same hosts that the zoonati claims to be “certainly the intermediate hosts for SARS-CoV-2”, speaks volume. They were also entirely incapable of emergence without engineering “push” as demonstrated by the near neighbors which all are FCS-free and spread just fine (even better than SARS-CoV-2) without it inside all manner of hosts. gab.com/Flavinkins/pos… In fact, the destruction of the Proline at 681 associated with VOC evolution in live hosts (human or animal hosts) simultaneously remove the virus’ ability to grow efficiently in laboratory cell lines: and the very weird and hard to explain lineages show evidence of reversion to culture adaptation. In the exact same time as illegal biolabs were found and when variants emerge without a traced location of origin or epidemiological link between cases. The Proline as it turned out is important for growth in VERO cells and variants that evolved in live hosts or with P681 mutated have defect in growth inside them. This mean that Wuhan is effectively the most VERO-suitable isolated variant over the course of the pandemic. gab.com/Flavinkins/pos… gab.com/Flavinkins/pos… https://gab.com/Flavinkins/posts/108682807199122313 archive.md/az10E archive.md/TrTW5 @mbw61567742 some of the features like HV6970 also show evidence of VERO association (P2V/HL6970). Not something that you expect for ZW, as the actual host it adapted to is VERO E6. In fact, all VOCs grow less efficiently in VERO E6 compared to non-VOC/“WT”. The same in the non-D614G A.23.1 strain as well. A striking graph below. https://gab.com/Flavinkins/posts/111398506038803573 Human or mice, the variants have less growth in VERO than Wuhan. Not something you expect for a virus that was not supposed to have seen a primate before the first human infection under the market theory. The FCS look exactly like a cell line adapted version after an insertion of the ENaC FCS as expected by DEFUSE during rescue and isolation—Both direct assembly and targeted RNA recombination are viable options for its insertion, and it is not unusual for a sample or a branch of its culture to be resequenced or deep sequenced months to years after sample collection and initial operations. The CGG-CGG is also not a coincidence—using it improve immunogenicity and allow efficient killed virus vaccine production and therefore adding a self leader failsafe for deployment, and manageability in case of unintentional release. Remember those HV6970/HL6970 (VERO adaptation of P2V).

@NestCommander - Kevin W. McCairn PhD
“Humanized mice will attenuate the FCS”=“humanized mice will generate the exact PRRAR site”. P681 and A372=VERO cells. And Q498=Mus Musculus germline immune system with human ACE2. Also reality: it was not “out of frame”. SARS-CoV-2 uniquely have two dS changes compared to all other QTQTNS genomes after the last Cysteine before the first S cleavage site. Shi put it in S2 And the Proline is so you can grow it into a stock in VERO E6 cells (VOCs or P681 mutants have growth defects in VERO cells) The PRRVR from mouse-passaged MERS-CoV.
@NestCommander - Kevin W. McCairn PhD
In fact, the Proline and the QTQTNS are really only stable in VERO and CaLu-3 (VERO/HAE) cells. In live hosts, P681 mutates to R681 or H681, and QTQTNS mutates to QTQTKS. There is no middle ground except if you still need to breed to stock quantity within VERO E6 cells.




@NestCommander - Kevin W. McCairn PhD
Bonus: for VERO/HAE cultures, deletions of the S1-S2 forms an equilibrium with QTQTNSPRRARS in ratios from 5% to ~70%. These mutations are actually identified within the Wuhan patients themselves, obtained from clinical samples in stead of only after culture for the first patients. Such clean QTQTN or SPRRARS deletions are not found even in homology in natural SARSr-CoVs. You only get to an FCS and the Proline (in stead of R, H or A) stabilized within these liquid medium-immersed cell cultures, and only if you start with an synthetically inserted FCS such as with the hENaC, the closest “human-specific cleavage site” to the QTQTNSRSVAS which “clear mismatches occur” at the first of the two S2 cleavages site in the Spike.




@NestCommander - Kevin W. McCairn PhD
https://gab.com/Flavinkins/posts/109640519028841414 It is not just that SARS-CoV-2 Wuhan grows best in VERO cells out of all variants. Some earliest patients harbored inside their QS specific S1-S2 deletions that can form only in VERO E6. https://gab.com/Flavinkins/posts/109888056517115303 And These seems to be related to cell surface expression that have most relevance to Spike-nanoparticles for use in non-humans. They are not vaccines that can be used in humans due to the human signal peptide used and the pcDNA3.1 which contained undesirable proteins. They are also not pseudoviruses. They best fit the “Spike nanoparticles” specified in DEFUSE out of all. (As a plain binding study would not use a complicated transmembrane anchor, which interfered with pseudovirus assembly. Human tpA signal peptide and pcDNA3.1 mean the formulation is unsafe for humans, which should not happen for such clearly finished-for-mass-production-in-HEK293f nanoparticle (that also have envelopes) formulations unless it is intended only for non-humans (such as DEFUSE bats).) https://t.co/gpv4cXu1WP. https://archive.md/1C7om

@NestCommander - Kevin W. McCairn PhD
In fact, the XRRXRX motif is considered a signature of cell culture adaptation, in stead of live host adaptation which the Heparan sulfate-binding motif is invariable broken. journals.asm.org/doi/10.1128/JV…




@NestCommander - Kevin W. McCairn PhD
Initially they do not have sufficient samples for an MRCA analysis, and that they were satisfied with only lineage B being available at the market. However, Eventually it was found out that lineage A is the more ancestral strain, and they have to make up a sample to put it into the market. https://archive.md/ANS4Q they came up with “A20”, inconsistent in both the ratio of 8782/28144 and in the ratio of reads vs Ct values with the other samples they claimed to show. The way they adulterated the post-26-03/2023 datasets is also one of the reason why the jbloom et al datasets gets humans as higher ranked in the alignments in the positive samples compared to all samples in both all sampling dates and Jan 12—They do it by dropping random human reads into the “negative samples” and scrambling the rest of the animal reads, all uploaded after 26/03/2023, resulting in an reduction of spread of correlation metrics over all species and correlatedness with humans for all samples compred positive samples only, not only in Jan 12 but for all sampling dates. In fact, all 3 samples that are different between 2021 and 2023 are also samples that have additional datasets uploaded in 26/03/2023 after an 03-10/03/2023 upload. Sample A20 have distinct host composition between the 03-10/03/2023 (without lineage reads) and 26/03/2023 upload, which is not expected from “viral amplicon sequencing” (with lineage A reads) which does not perturb the host reads if genuinely from the same sample. This is consistent with the general scrambling of host sequences within the “post-26/03/2023” samples, and showcases irreconcilable dishonesty within this sample set especially when sample B5, likely used as a standard, remain unchanged, creating an additional inconsistency in term of protocols—one set of numbers in 2021, one set of numbers in 03/03/2023, and a third set of numbers in 26/03/2023. Zero custody in the CCP’s grasp up to upload, change constantly per demand of the leading zoonosis theory—“change those ‘data’ on the fly, based on any reactions and feedback, make up your uploads to attempt pushing zoonosis as hard as possible”. This is also why the mutual information between SARS-CoV-2 and any species at all, especially all land-dwelling species, are completely destroyed upon inclusion of the 26/03/2023 upload date. These are the species that they seek to scramble reads in order to remove the correlational edge of Homo Sapiens that have remained despite their attempt at filtering their previous “data”. In Mar-Apr 2020, China officially blamed wild animals sold in the Huanan market. Publishing the “data” as currently seen to Holmes would be the best way to solidify this then-official opinion. If the “market environmental data” can be interpreted in any way to arrive at the C-C “conclusions”, ECH won’t be denied of it. Since he is denied, the most logical reason for the denial is that it does not originally support any of the C-C “conclusions”, and were tampered only recently to poison the scientific database and to provide a fallback for debate purposes. Only after evident in-vitro and in-silico tampering and subsequent approval by the CCP, would it be officially permitted—in fact, actively given to Holmes for “analysis”. Despite attempts at scrubbing all 300nt+ non-viral human Contigs from the “positive wildlife stall samples”, which have led to an inverse correlation between the 300nt+ contigs left inside these samples and the product of Homo Sapiens and SARS-CoV-2, mutual information and the ratio between the leftover human mitochondrial reads and SARS-CoV-2 have been preserved as the removal process preserved ratios, And you still end up with Homo Sapiens being the most mutually informant species for SARS-CoV-2 whenever significant mutual information is preserved at all within a slice of the “dataset”.




@NestCommander - Kevin W. McCairn PhD
In fact, there are even further inconsistencies in these so-called “data releases” which further indicate that there are both sufficient number of unaccounted flow cells to source all the reads necessary for scrambling the post-28/03/2023 “datasets” and that there are likely both selective representation of and cannibalization-and-redistribution of sample datasets, with once again a total absence of custody information or in deed, from what mix of material was the actual source libraries constructed from, suggested that the CCP used a strategy of “holding back as many reads as that would be needed to adjust the datasets to whatever direction to promote the arrival at a “likely zoonosis” conclusion so there is always a conclusion to jump to if the lab is indeflectably blamed” when posting the “dataset” or the associated publications—not even the number of samples per category could be matched to the percentages published. (see how the mutual information metric, e.g. the plausibility to use regression from the other points on the correlation graph to identify the location of a dot on the graph from only one axis of its coordinates e.g. the ability to predict the concentration of virus from species and vice versa, are completely crashed upon inclusion of the “26/03/2023” samples in both Jan 12 and all collection dates)


@NestCommander - Kevin W. McCairn PhD
https://archive.md/VNr75 In fact, contrary to the claim that they are “near the market”, all 3 of the lineage A samples in Early Wuhan was actually found in tight and direct linkage with the WCDC much more than and as opposed to the Huanan market. One is A20, the WCDC sampler PPE which would be among the first sample to replace outright in stead of merely filtering and scrambling to minimize leak of lab-linked information. Exactly what to make it for best fit to the running theory, however, they stumbled and changed twice with feedback and changing demands, resulting in three distinct and dishonestly-inconsistent-with-each-other datasets and results. Another stayed in a Hotel, which is right next-door to the “new” WCDC site where samples and cultures would have been transported to and workers would move back and forth between when they have just finished setting up the new lab and have started experimentation which needed materials likely exist in both the old and the new site at the beginning of its operation. The third, “cluster 1”, is right on the route of this back and forth commutation and near the “old” WCDC site, “somewhere near the Xinhua hospital”. All have strong linkage to the WCDC and none documented credible linkage to transmission at the Huanan market. The WCDC and the Hubei CDC stores all of the human samples and backups of research cultures of pathogenic microbes in Wuhan, as this is their legally delegated duty (the “各级疾控部门” are termed “保藏机构” for “病原性微生物” under Chinese law governing the use of cultures and samples of human and animal pathogenic microbial samples, and samples that were suspected to have the possibility of containing such microorganisms. These are also the only locations which first round samples arriving in Wuhan are allowed to go for pre-screening prior to entry into the other labs in Wuhan, “检测机构”. ) and that labs in China are not allowed to store such cultures except several select state key laboratories. Since 2014, the only EID surveillance target in Wuhan is the HSM which all other sites are kept blind so that they can blame Huanan in case the research labs suffer an accident. It is likely that the WCDC (but not the Hubei CDC) would internally get the wind of an “SARSr-CoV” (with an antigen kit that were apparently available to many high-level hospitals in Wuhan) almost the soon their surveillance program is tripped in 20-22/12/2019 with their first hospital-visited market case. After an initial release from the WIV that caused Chen’s infection, and eventual transmission to the HSM via line 2 of the Wuhan metro, they mobilized the WCDC in 20-22/12/2019 to begin tapering with the environmental samples (largely based on the leading zoonosis theory proposed or identified at that time) and prepare for any needed scapegoat action. That mobilization ended up causing an infection of a WCDC worker with an aliquot of a sample containing WA1, A and B in the same quasispecies, which then go on infecting all of the earliest lineage A cases in Wuhan.
@NestCommander - Kevin W. McCairn PhD
The WHO report is found to have moved all https://gab.com/Flavinkins/posts/109048819612838694 cases with onset before 27/12/2019 https://www.researchgate.net/publication/370635299_Greater_than_the_Sum_of_its_Parts_-_Aggregated_Wuhan_COVID-19_case_data_points_to_the_wrong_side_of_the_Yangtze_River_-_Rixey_-_20230509 inside the district of Wuchang, and then all cases within the central Wuchang prefectures and those prefectures near the labs, to Jianghan. with calibration performed so that they would add up to a perfect “bullseye” https://www.reuters.com/article/us-health-coronavirus-who-china-idUSKBN2AD090 https://www.nytimes.com/2021/02/12/world/asia/china-world-health-organization-coronavirus.html https://www.washingtonpost.com/opinions/2022/11/17/covid-early-cases-wuhan-china-mystery/ https://www.wsj.com/amp/articles/china-refuses-to-give-who-raw-data-on-early-covid-19-cases-11613150580 of within 50m precision at the Huanan market, which would not be realistically possible given the expected social biases from uneven residential densities even in the neighborhood of the Huanan market. Cases *residences* were dropped into water and placed into non-residential areas as the result of this tampering, especially the former which, residences could not exist on since they would be washed away by the water. Obvious examples included accountant Chen, which they refused to mention where he lived at all since the WHO report alongside any specific single cases, and one of the two “江夏急救中心” ambulances seen blaring into Wuchang in 31/13/2019, where only one out of the two, one that likely did not live in Shidong, were counted as a dot in Jiangxia. They refused to give any line lists at all for a reason.
@NestCommander - Kevin W. McCairn PhD
Tampering with data, moving cases, contradicting own early media reports, this is the reality of that “market centered” early cases “dataset”. The “dataset” is as fake and as inconsistent as a $3 bill. https://gab.com/Flavinkins/posts/109256201942085712 They completely eliminated all cases that landed within the borders of the Wuchang district before 27/12/2019, to the point that they claim to see cases further east of but not within Wuchang. This is the cluster that they were hiding. Again, the early cases are WA1, which even when cluster, failed to meet the recognition requirements. https://gab.com/Flavinkins/posts/109248812361151175 The “中部战区总医院” reported that they were taking in 700+ cases in a single day at 31/12/2019 all in the fever clinics. https://gab.com/Flavinkins/posts/109400051863347812 Then there is a media coverage bias.




@NestCommander - Kevin W. McCairn PhD
Myanmar, Cambodia, which also, incidentally, was where China specifically sampled before. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7818139/ This also included human sampling as well, per “pathogen host adaptation and immune intervention”. Now where I find these specific human Mitochondrial haplotypes again? (Southeast Asia, not central China, +SARS-CoV-2, no FCS, exactly where the WIV and the WHU would sample humans and put the resulting cultures into the WCDC). Note how the China serological sampling is a specific sampling around bat caves which human cells-infecting CoVs have been specifically isolated before. Notice how low this number is compared to southeast Asia (Cambodia, Myanmar). Despite Shi and Daszak’s name on it, “no work was ever conducted in Laos” they claimed. How can the EHA be trusted?




@NestCommander - Kevin W. McCairn PhD
Not only Leaked SRA data included both the exact kind of viruses that they claim will not be present in the WIV—and the exact SARS-CoV-2, WA1, cultured in a CoV-specific tailored fusion cell line VERO-CHO never used in China and sequenced before even a sample of WA1 can be taken in China, alongside C/C and B, at high passage depths, and contained within it residual human DNA not from anywhere in central China but in stead right where they were sampling from the 2018 “pathogen host adaptation and immune intervention” grant—the belt and road regions; https://gab.com/Flavinkins/posts/109888056517115303 But also these membrane anchored cellnsurface expression vectors intended for HEK293f of the SARS-CoV-2 Spike that have most relevance to Spike-nanoparticles for use in non-humans. They are not vaccines that can be used in humans due to the human signal peptide used (expressing an antigen together with a human protein, especially when co-localized through generation of nanoparticles processed from the same peptide chain) and the pcDNA3.1 which contained undesirable proteins. They are also not pseudoviruses. They best fit the “Spike nanoparticles” specified in DEFUSE out of all. (As a plain binding study would not use a complicated transmembrane anchor, which interfered with pseudovirus assembly. Human tpA signal peptide and pcDNA3.1 mean the formulation is unsafe for humans, which should not happen for such clearly finished-for-mass-production-in-HEK293f nanoparticle (that also have envelopes) formulations unless it is intended only for non-humans (such as DEFUSE bats).) https://t.co/gpv4cXu1WP. https://archive.md/1C7om Continued EHA human sampling=Yunnan and belt and road DNA. Isolate if possible=special unpublished VERO-CHO cells. And it was sequenced before the first public sequencing of SARS-CoV-2 with this machine type by the flow cell, confirmed via Sangon policy and Chinese law, and before+not matching any samples of WA1 was even taken in China. And this exact CAS special project mirroring of DEFUSE+Year 5 extension—sample humans from belt and road area, isolate and engineer viruses for infection characterization, and create vectorized and nanoparticle vaccines that are capable of bringing in both backbone and Spike into bats studied in and released by the WIV, and into the main sample storage facility of the WCDC. (Also see this—note all the FCS relevant oddities can also be caused by targeted RNA recombination link.springer.com/chapter/10.100… followed by cell culture). The instability associated with 8782/2814/18060 (WA1->A->B) is found to recur at least 3 times in the WA1/UW cluster, especially their cultured isolates. The associated samples have T22657C, T3346C, A21562C and G487T. all of which is in RaTg13 but not in WuHu-1. also T1963C and T22963C in BANAL-52. https://gab.com/Flavinkins/posts/109640519028841414 It is not just that SARS-CoV-2 Wuhan grows best in VERO cells out of all variants. Some earliest patients harbored inside their QS specific S1-S2 deletions that can form only in VERO E6.
@NestCommander - Kevin W. McCairn PhD
So, where are DEFUSE going to sample humans? Also, the RaTg13 RBD bind human ACE2 poorly, resulting in exceptionally high sVNT cross-reactivity as even poorly binding antibodies can display ACE2 off it. Once again, 1: RaTG13 is not viable. https://zenodo.org/record/5702700#.ZJ2KiyV6slT https://zenodo.org/record/5778318#.ZJ5hyCV6slT 2: the real issue is that 1. WIV lies about everything serological. None of their “tests” were positive when politics require it to be negative. And 2. The missing sequences of Latinne et al is where you find what the WIV was working on. 7 SARSr and 54 total CoVs were missing entirely.




@NestCommander - Kevin W. McCairn PhD
Note how the WHU itself gets about half of all the animal work that involved the “understanding risk of bat coronavirus emergence” grant— Both it and DEFUSE are included in the “pathogen host adaptation and immune intervention” grant.
@NestCommander - Kevin W. McCairn PhD
@NestCommander - Kevin W. McCairn PhD
And in deed, this 2018 EHA grant approval document invoked an near exact replication of DEFUSE on what to do with the new SARSr-CoVs—gather Spikes 10%-25% divergent from SARS-CoV-1, test with chimeric viruses first, and then take the immune-evasive and human infectious ones, and validate with full genomes—clearly more than “only 1-2” with binding to human cells first, see SARS-like signs from some of these next, and then some (more than 1 but nor all) don’t respond to mAbs, vaccines, etc.




@NestCommander - Kevin W. McCairn PhD
@BlackTomThePyr8 - Tom Czerniawski
Few will ever understand the gravity of what Lt. Col. Murphy did. By releasing DEFUSE he showed us where to look. From that discovery, subsequent draft DEFUSE documents were discovered years later by @USRightToKnow that proved C-19 to be synthetic, and revealed how it was made.




@NestCommander - Kevin W. McCairn PhD
What those analyzers think: “many different precise and specific pieces recombine into SARS-CoV-2”. Reality: an unpublished but readily sampled SARS-CoV-2 progenitor spread fragments over time into multiple locations. Some end up in the published samples. “10%-25% Spike divergence”—they will isolate RBDs from viruses that they sample to identify one that bind ACE2. Only fine tuning is needed later. “Exotic recombination”? It is just reverse transcriptases in cases of “postpandemic inserts”. How many billion hosts are needed and how this compare to the total number of wild animals on the entire Earth? link.springer.com/chapter/10.100… Use targeted RNA recombination if you have a cultured virus in stead. “Perfect synergy”? Nothing but VERO/HAE cells. Those “synergy” are only stable here. Not in any live hosts which they will mutate and destroy each other in stead. https://gab.com/Flavinkins/posts/109205261283826972 ReCCA is tautological and fictitious. https://gab.com/Flavinkins/posts/109863181504837302 https://gab.com/Flavinkins/posts/109465063042828622 https://gab.com/Flavinkins/posts/109255356915252021 BtSY2 is sequenced in 2018. https://gab.com/Flavinkins/posts/109399710986742685 BANAL is in the hands of the DOD in 2017. https://gab.com/Flavinkins/posts/109340247585238829 https://gab.com/Flavinkins/posts/109800300869616862 And all of which got funneled into the EHA, which eventually will end up in the WIV.


@NestCommander - Kevin W. McCairn PhD
Before they begun enforcing their claim of “100/174 centered around the market” and starting to tamper with data to make the claim, https://ghrp.biomedcentral.com/articles/10.1186/s41256-021-00200-8 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7149375/ 135/92 and 115/82 cases already got into in early peer-reviewed papers that went missing in the WHO report. Past media reports archive.md/Ea0Kw archive.md/1x658 also contradict WHO in key early cases’ residences, including the earliest case they admit in the WHO report. archive.md/5sdkR archive.md/1pcCU archive.md/N0hib archive.md/VXtu9 archive.is/Kyr1z https://archive.org/details/mace-e-pai-covid-19-analysis-redacted/page/8/mode/1up And you know that they hate this information when it was censored. The MACE-EPAI document here is not searchable on google. Up to one third of all cases were either removed completely or moved toward the market in the “dataset”. archive.md/zUD1F archive.md/Pc6gp https://archive.is/p3K3Z Including the very first case they ever admitted officially. And outright removed 4 times more cases than official. Unlinked cases supposedly secondary to linked cases should cluster around them, not the market itself. archive.md/GvRcD archive.md/ZgVzp Wuhan authorities after that archive.md/OIGPz 2014 incident now targeted only the Huanan market when looking for EID outbreaks—and nowhere else. archive.md/1x658 They tampered with the early cases data archive.md/Ea0Kw To make it look like it “started at the market” when in reality the first case they ever admitted lived right next to the WIV BSL-4. archive.md/5sdkR severe discrepancy happening December 2019 and January 2020 indicate tampering with case counts. archive.md/1pcCU This is indicative of catastrophic ascertainment bias was going on. None of China’s “early cases” dataset is credible. https://archive.md/ET1GA https://archive.md/Ea0Kw https://archive.md/1x658 The tampering of early case residence data is systematic and extensive. It is the reason why they refused to provide this data in any detail at all. Not only did The first every case they admitted live in Shidong right next to the BSL-4, and were moved toward the market in the WHO report in contradiction to all known media coverage, https://gab.com/Flavinkins/posts/109256201942085712 the entirety of Wuchang district was wiped clean for every single WHO case that have onset before 27/12/2019–with up to 3000 cases moved to the market this way over the entire Wuhan outbreak. https://archive.md/1x658 and for central Wuchang near the labs and the densest inhabited regions inside the district, all cases were moved away in the WHO map. Unfortunately Rasmussen's work on the origins question rests heavily on what David Relman described as "hopelessly impoverished" early case data. https://www.washingtonpost.com/national-security/2023/02/27/little-known-scientific-team-behind-new-assessment-covid-19-origins/ https://www.washingtonpost.com/opinions/2022/11/17/covid-early-cases-wuhan-china-mystery/ https://archive.md/ke1lp https://archive.md/RaYPC David Fisman: I think the most interesting thing this fellow says is that there are clearly tens of thousands of cases...That implies a much earlier introduction than would have occurred with a seafood market outbreak..."
@NestCommander - Kevin W. McCairn PhD
https://www.nytimes.com/2021/02/12/world/asia/china-world-health-organization-coronavirus.html https://archive.md/UFrSv They systematically moved more than 3000 cases from the lab to the market and gave “cases data” that they wanted to push for market as first outbreak site to distance from the labs. https://www.researchgate.net/publication/370635299_Greater_than_the_Sum_of_its_Parts_-_Aggregated_Wuhan_COVID-19_case_data_points_to_the_wrong_side_of_the_Yangtze_River_-_Rixey_-_20230509 Such an result of having unlinked cases closer to the market than linked cases is not expected even under the null hypothesis of market origin, which we should see unlinked cases secondary to and cluster around the linked cases, and not the market itself. https://www.researchgate.net/publication/370635299_Greater_than_the_Sum_of_its_Parts_-_Aggregated_Wuhan_COVID-19_case_data_points_to_the_wrong_side_of_the_Yangtze_River_-_Rixey_-_20230509 Not only there were an complete absence of verifiability in Chinese cases, there is direct non-circumstantial evidence that they moved up to 3000 cases from Wuchang to Huanan. In fact, it is totally not normal to have unlinked cases closer to the market than linked cases—the only way this can happen is with ascertainment bias. Only near the market gets ascertained if not directly linked to it. Base rate neglect. They did the exact same thing when claiming that all 67 “pre-Huanan checkable cases” were “serologically negative”. Again, the social media associated here say “before Jan 18, 2020”. Included all Dec cases. https://www.mdpi.com/2220-9964/9/6/402 It is actually impossible for unlinked cases, supposedly secondary, to cluster closer to the market than linked cases which supposedly to be primary, without significant sampling bias or outright manipulation in the underlying “data”. Both evidently happened. https://arxiv.org/pdf/2401.08680.pdf https://archive.md/JVFuc If you toss away anything that is not officially announced by China in bold, then obviously you would arrive at exactly what China wanted you to believe.
@blink64 - polyploidy
When actual statisticians have a go at your paper purporting to show the SARS2 outbreak originates at the HSM. Thanks @gdemaneuf! https://t.co/avfBP4bZPa

@NestCommander - Kevin W. McCairn PhD
@threadreaderapp unroll
@NestCommander - Kevin W. McCairn PhD
https://twitter.com/vigilantfox/status/1686198223763853312 Fauci lied under oath “not funding gain of function (GOF) research” https://twitter.com/charlesrixey/status/1728099199772762618 Email: https://twitter.com/texaslindsay_/status/1679838118743097348 GOF under another name: https://twitter.com/s_q_e_r_l/status/1392429082370002946 Even under their own standards for published results. https://twitter.com/r_h_ebright/status/1704641787380298152 Fauci directly offshore GOF. https://twitter.com/hansmahncke/status/1752063214769377292 https://twitter.com/r_h_ebright/status/1633624833081884674 https://twitter.com/schwinn3/status/1457368942272565255 Another discrepancy between public propaganda plans and private concerns. https://twitter.com/covidselect/status/1730198241428275213 Flip-flop with masks is the least of his worries. https://twitter.com/covidselect/status/1730198245626769905 See replies for his past lies. https://twitter.com/covidselect/status/1730198247971373554 More about Fauci: https://twitter.com/covidselect/status/1732078538033971626 “Fauci's legacy is as honorable as this NIAID report he was obligated to release under a FOIA lawsuit is “packed with information”...” https://twitter.com/williewwilliam/status/1730356526424932620 Fauci lied, people died. https://twitter.com/covidselect/status/1730198247971373554 FOIA stuff: https://twitter.com/thackerpd/status/1412369872487698432 Yes. They are really there defending GOF research and their grave COI making them wholly opposed of even the idea that a pandemic can ever be initiated by virology research. http://bmj.com/content/344/bmj.e2398/rapid-responses… https://twitter.com/daoyu15/status/1712998816356643194 You can only have GoF work generate a viable prediction or a vaccine if the next emergence is of the exact same genetic makeup as your GOF strain. The extreme diversity of viruses mean that you have (total number of viruses in the world (billions)*probability that a GOF study result being used maliciously (>1/200 minimum)) times higher likelihood that GOF research cause a pandemic in stead of preventing it. http://archive.ph/BToZR http://archive.md/YYIXp http://gab.com/Flavinkins/posts/109663743902085653… https://twitter.com/daoyu15/status/1713099142841729200 https://twitter.com/Daoyu15/status/1725182075656155186 The ODNI both broke the law and is full of elementary mistakes in their “report”. That is why it can’t be trusted. https://twitter.com/daoyu15/status/1725274033103491368 As on why? https://twitter.com/daoyu15/status/1725276084965380325 They need to protect GOF at all costs. https://twitter.com/daoyu15/status/1725182099232305536 More hidden experiments. https://twitter.com/0rf/status/1594467737744465920?s=46&t=wRQSWp_1VffWmS2vKQwhSA… Covid origin declassification act violation of the ODNI. https://twitter.com/daoyu15/status/1724626293403373877?s=46&t=wRQSWp_1VffWmS2vKQwhSA… "Former Acting Assistant Secretary of State Thomas DiNanno tells [Sky News]…that when his team unearthed explosive evidence that pointed to a laboratory leak…, the intelligence community ran interference in support of a natural origin narrative." https://twitter.com/r_h_ebright/status/1729164212159824154?s=46&t=wRQSWp_1VffWmS2vKQwhSAw… GOF is only for dual-use bioweapons research and are never useful for any kind of vaccine or therapeutics. Stop lying. https://twitter.com/Daoyu15/status/1682701118655303680 https://twitter.com/daoyu15/status/1679461360164552705 Here is clearly KGA never ruling out even engineering. https://twitter.com/daoyu15/status/1690480261103034368 And unfortunately, the first use of the prefusion-stabilized Spike was to target MERS-CoV and WIV1 wasn’t the drive of the patent, nor did the patent required any GOF research. Also, the vaccines are now proven to be hazardous, driving unending waves of reinfections. https://twitter.com/daoyu15/status/1691993269847413159 https://twitter.com/daoyu15/status/1691993693258186857 https://twitter.com/daoyu15/status/1724491689124065595?s=46&t=wRQSWp_1VffWmS2vKQwhSA… Not even Tedros consider their claimed conclusions valid. https://twitter.com/drtedros/status/1724132394662252877?s=46&t=wRQSWp_1VffWmS2vKQwhSA… Unanimously, they passed a law banning all NIH ePPP GOF funding. https://twitter.com/justinrgoodman/status/1724621384305742069?s=46&t=wRQSWp_1VffWmS2vKQwhSA… Numerous problems of the ODNI “report”. https://twitter.com/daoyu15/status/1724626293403373877?s=46&t=wRQSWp_1VffWmS2vKQwhSA… https://twitter.com/daoyu15/status/1738419581080010945 Red-faced and looking extremely anxious, Dr. Peter Daszak, President of EcoHealth Alliance, testified today in a closed hearing about the #OriginOfCovid. https://twitter.com/daoyu15/status/1725480788861559101 Following his testimony, the House of Representatives unanimously voted to defund two active grants awarded to him. https://twitter.com/fermentillc/status/1724744680183611631 https://twitter.com/drhermiz/status/1724620221464424608 https://twitter.com/justinrgoodman/status/1724597207741902969 Including additional approved bills that cuts all DOD funding to the EHA as well. https://twitter.com/Bryce_Nickels/status/1730668384583299361 Evidently, the secret bioweapons department of the U.S. wasn’t happy on this resolution. https://twitter.com/BlackTomThePyr8/status/1730674382580678812 But even they agreed that bat CoV GOF work involving China as a region or using the WIV is too risky to continue putting DOD money on. https://twitter.com/justinrgoodman/status/1732740769071423590 https://twitter.com/daoyu15/status/1732967051617288443 https://twitter.com/daoyu15/status/1733530016695386345 Like touching a fire and got burned. https://twitter.com/daoyu15/status/1732968216706842766

@NestCommander - Kevin W. McCairn PhD
https://gab.com/Flavinkins/posts/108971775263920617 Massive status bias in peer review

@NestCommander - Kevin W. McCairn PhD
The Pan-Zootic Super-PRION: An Ancient Extinction Level Event? https://t.co/J1AGUnbEnH
@NestCommander - Kevin W. McCairn PhD
Robert you had the chance over the last 2.5 years to be talking about the amyloidogenic/prionergic properties of HIV GP120. Instead you've been kvetching about not being credited for LNP & mRNA technologies. An opportunity missed to inform the public about the real issues around biowarfare and weaponization prion/amyloidogenic catalyzing epitopes. Here is an example of what you should be talking about, if you need help with the neuroscience you just have to ask. https://rumble.com/v3w8rjx-savims-toxic-peptides-amyloids-and-prions-from-genetic-vaccines-to-sars-cov.html @Doctor_I_am_The @Jikkyleaks @Kevin_McKernan @CharlesRixey
